
Крупнейшая бесплатная
информационно-справочная система онлайн доступа к полному собранию технических нормативно-правовых актов
РФ. Огромная база технических нормативов (более 150 тысяч документов) и полное собрание национальных стандартов, аутентичное официальной базе Госстандарта.
GOSTRF.com — это
более 1 Терабайта бесплатной технической информации для всех
пользователей интернета. Все электронные копии представленных здесь документов могут распространяться без каких-либо ограничений. Поощряется распространение информации с этого сайта на любых других ресурсах. Каждый человек имеет право на неограниченный доступ к этим документам! Каждый человек имеет право на знание требований, изложенных в данных нормативно-правовых актах!
| Поддержать проект |
| Скачать базу одним архивом |
| Скачать обновления |


Инструкция по лабораторному контролю очистных сооружений на животноводческих комплексах
| Статус: | не действует |
| Название рус.: | Инструкция по лабораторному контролю очистных сооружений на животноводческих комплексах |
| Дата добавления в базу: | 01.09.2013 |
| Дата актуализации: | 01.01.2021 |
| Область применения: | Инструкция включает правила организации лабораторий по контролю за работой очистных сооружений, методики отбора и хранения проб жидкого навоза и продуктов его переработки, проведение химических, санитарно-бактериологических и гельминтологических анализов, методы оценки результатов контрольных измерений, правила техники безопасности при работе в лаборатории и предназначена для использования в лабораториях действующих и строящихся комплексов по производству животноводческой продукции. |
| Оглавление: | Часть I Часть II Часть III |
| Разработан: | ЛИСИ Минвуза РСФСР ВИГИС ГипроНИсельхоз (Институт Гипронисельхоз ) НИПТИМЭСХ Росгипрониисельстрой Свинопром РСФСР ВНИИВС |
| Утверждён: | 17.11.1980 Министерство сельского хозяйства СССР (USSR Ministry of Agriculture ) |
| Принят: | Минздрав СССР (USSR Minzdrav ) Главсельстройпроект Минсельхоза СССР (Glavselstroyproject, USSR Minselkhoz ) Главживпром СССР (USSR Glavzhivprom ) |
| Расположен в: | Техническая документация Экология СЕЛЬСКОЕ ХОЗЯЙСТВО Земледелие и лесоводство Животноводство и селекция животных |
| Нормативные ссылки: |
|

 «Часть I. Организация лаборатории. Методы санитарно-бактериологического и гельминтологического анализа сточных вод»
«Часть I. Организация лаборатории. Методы санитарно-бактериологического и гельминтологического анализа сточных вод»Скачать
Содержание
- Правила. Ветеринарно-санитарные правила подготовки к использованию в качестве органических удобрений навоза, помета и стоков при инфекционных и инвазионных болезнях животных и птицы
- Часть 1. Общие положения
- Часть 2. Обеззараживание навоза, помета и стоков
- Часть 3. Контроль обеззараживания органических удобрений
- Часть 4. Хранение и транспортирование
- Часть 5. Использование навоза и навозных стоков
- Приложение 1. Методики подготовки проб органических удобрений и исследование их на наличие индикаторных микроорганизмов
- Приложение 2. Питательные среды
- Приложение 3. Виды навоза и способы его обеззараживания
- Приложение 4. Максимальные сроки выживаемости возбудителей инфекционных болезней во внешней среде
| Утверждаю
Заместитель Начальника Департамента ветеринарии Минсельхозпрода России В.В.СЕЛИВЕРСТОВ 4 августа 1997 г. N 13-7-2/1027 |
Правила
Ветеринарно-санитарные правила подготовки к использованию в качестве органических удобрений навоза, помета и стоков при инфекционных и инвазионных болезнях животных и птицы
Часть 1
Общие положения
1.1. Ветеринарно-санитарные правила подготовки к использованию в качестве органических удобрений навоза, помета и стоков животноводческих и птицеводческих предприятий, именуемые в дальнейшем «Правила», предназначены для осуществления контроля за проектированием, строительством и эксплуатацией сооружений подготовки навоза, помета и стоков, с целью получения экологически безопасных органических удобрений, обеспечивающих охрану окружающей среды от загрязнений возбудителями инфекционных и инвазионных болезней.
1.2. «Правила» подготовлены на основании законодательных и нормативных документов:
- Закон Российской Федерации «О ветеринарии» от 14 мая 1993 г. N 4979-1;
- ГОСТ 24076-84 «Навоз жидкий. Ветеринарно-санитарные требования к обработке, хранению, транспортированию и использованию»;
- «Общесоюзные нормы технологического проектирования систем удаления и подготовки к использованию навоза», ОНТП 17-86, Госагропром СССР;
- «Республиканские нормы технологического проектирования птицеводческих предприятий», РНТП 4-93;
- «Инструкция по лабораторному контролю очистных сооружений на животноводческих комплексах», 1980 (МСХ СССР);
- «Инструкция по проведению ветеринарной дезинфекции объектов животноводства», 1989 (Госагропром СССР);
- «Ветеринарно-санитарные и гигиенические требования к устройству технологических линий удаления, обработки, обеззараживания и утилизации навоза, получаемого на животноводческих комплексах и фермах», 1979 (МСХ СССР, Минздрав СССР);
- «Методические рекомендации по предотвращению загрязнения окружающей среды бесподстилочным навозом животноводческих комплексов и ферм», 1989 (Госагропром СССР и Госкомприроды СССР);
- «Оросительные системы с использованием животноводческих стоков. ВСН 33-2.2.01-85» (Министерство мелиорации и водного хозяйства СССР);
- «Ветеринарно-санитарные правила по использованию животноводческих стоков для орошения и удобрения пастбищ», 1993 (Минсельхоз России, Департамент ветеринарии);
- ТУ 10-11-887-90 «Компост торфонавозный из навоза крупного рогатого скота»;
- ТУ 64-4688624-02-91 «Вермикомпост».
1.3. Настоящие «Правила» распространяются на все виды органических удобрений, получаемых на существующих, вновь строящихся и реконструируемых животноводческих предприятиях различной мощности.
1.4. Выбор систем очистных сооружений подготовки органических удобрений проводят на основании технико-экономического сравнения различных вариантов с учетом специализации и типоразмера предприятия, климатических, почвенных и гидрогеологических условий.
1.5. Проекты систем обработки, хранения и обеззараживания органических удобрений подлежат согласованию с местными органами госветнадзора, госсанэпиднадзора и Госкомприроды.
1.6. При выборе места для строительства животноводческих объектов и птицефабрик необходимо предусматривать выделение сельскохозяйственных угодий для утилизации всего годового объема органических удобрений либо технологии переработки, обеспечивающие уменьшение объемов получаемых удобрений.
1.7. Сооружения подготовки навоза, помета и стоков располагают за пределами ограждений территорий ферм, комплексов и птицефабрик с подветренной стороны и ниже водозаборных сооружений.
Расстояние от сооружений до жилой застройки и животноводческих помещений зависит от мощности предприятий и определяется по таблице 1.
| Сооружения | Расстояние в метрах | |
|---|---|---|
| от животноводческих зданий | от жилой застройки | |
| Сооружения механической и биологической обработки жидкого навоза на фермах и комплексах | ||
| а) свиноводческие: — менее 12 тыс. в год | не менее 60 | не менее 500 |
| — 12 — 54 тыс. в год | не менее 60 | не менее 1500 |
| — 54 в год и более | не менее 60 | не менее 2000 |
| б) крупного рогатого скота: — менее 1200 коров | не менее 60 | не менее 300 |
| — 1200 — 2000 коров и до 6000 голов молодняка | не менее 60 | не менее 500 |
| — при больших размерах комплексов | не менее 60 | не менее 1000 |
| — открытые площадки на 10 — 30 тыс. голов | не менее 200 | не менее 3000 |
| в) овцеводческих на 5 — 30 тыс. голов | не менее 200 | не менее 3000 |
| Открытые хранилища (накопители) | ||
| — жидкого навоза | не менее 60 | не менее 1200 |
| — помета | не менее 200 | не менее 3000 |
| Биопруды и хранилища биологически обработанных стоков | ||
| не менее 60 | не менее 500 | |
| Площадки подготовки компостов малых ферм | ||
| — поголовье менее 50 голов | не менее 3 -5 | не менее 100 |
1.8. Все сооружения и строительные элементы систем подготовки органических удобрений должны быть выполнены с гидроизоляцией, исключающей фильтрацию жидкого навоза и стоков в водоносные горизонты и инфильтрацию грунтовых вод в технологическую линию.
1.9. Территория сооружений для подготовки органических удобрений должна быть ограждена, защищена многолетними зелеными насаждениями, благоустроена и иметь проезды и подъездную дорогу с твердым покрытием шириной не менее 3,5 м.
1.10. При разработке проектов сооружений следует предусматривать возможность карантинирования всех видов навоза и стоков в течение не менее 6 сут., необходимых для уточнения диагноза в случае подозрения на инфекционную болезнь.
Для карантинирования подстилочного навоза и помета сооружают площадки секционного типа с твердым покрытием, карантинирование бесподстилочного навоза осуществляют в специальных карантинных емкостях очистных сооружений либо в секциях навозонакопителей.
Хранилища для жидкого навоза оборудуют устройствами для перемешивания массы, скосы и днища их должны иметь твердое покрытие, закрытые хранилища необходимо оснастить люками, а также приточно-вытяжной вентиляцией.
При искусственной биологической очистке жидкого свиного навоза и сточных вод птицефабрик в аэротенках и последующей передаче их на городские очистные сооружения или сбросе в поверхностные водоемы карантинирование осуществляют с учетом времени пребывания их на очистных сооружениях предприятий.
Если в течение 6 сут. не зарегистрированы инфекционные болезни животных, навоз, помет и стоки обрабатывают по принятым технологиям, очищенные сточные воды сбрасывают в поверхностные водоемы в соответствии с требованиями «Санитарных правил и норм охраны поверхностных вод от загрязнения» (N 4630-88).
Часть 2
Обеззараживание навоза, помета и стоков
2.1. На случай возникновения инфекционных болезней животных на каждом животноводческом предприятии и птицефабрике должны быть предусмотрены способ и технические средства для обеззараживания навоза, помета и стоков. Продолжительность карантина в неблагополучных хозяйствах определяется действующими инструкциями о мероприятиях по ликвидации конкретных инфекционных болезней с учетом способа обеззараживания органических отходов, наличия дезинфектантов и технических средств, а также вида и устойчивости возбудителя болезни.
2.2. При возникновении инфекционных болезней в хозяйствах всю массу получаемых в этот период органических удобрений обеззараживают до разделения на фракции биологическими, химическими или физическими способами. Методы дезинфекции органических отходов следует предусматривать с учетом их физико-химических свойств, перспективных технологий обработки и возможности использования в качестве удобрений (Приложения 3, 4).
Для дезинвазии навоза, в особенности свиного и его смесей с другими видами навоза и помета, в целях уничтожения социально опасных возбудителей паразитарных болезней предусматривают соответствующие методы его обработки в системе удаления, хранения и утилизации. Одним из наиболее доступных является метод биотермической обработки в процессе хранения при определенных режимах.
2.3. Для свиноводческих комплексов мощностью 12 — 27 тыс. голов в год предусматривают проводить карантинирование в течение 6 сут. и обеззараживание от неспорообразующей патогенной микрофлоры неразделенного навоза путем длительного в течение 12 мес. выдерживания в секционных накопителях, анаэробной ферментацией в биоэнергетических установках или химическими средствами в карантинных или специально предусмотренных емкостях.
Биологический метод дегельминтизации также предусматривает выдерживание полужидкого и жидкого навоза свиней в открытых навозохранилищах в течение 12 мес.
Дегельминтизацию жидкой фракции свиного навоза осуществляют способом отстаивания ее в течение 6 сут. в секционных прудах-накопителях, оборудованных устройствами, исключающими попадание донного осадка в оросительную систему, и устройствами, обеспечивающими периодическую выгрузку осадка перед новым заполнением их жидкой фракцией.
2.4. Анаэробная ферментация жидкого свиного навоза осуществляется в биоэнергетических установках (БЭУ). Применение комплектов оборудования для анаэробного сбраживания возможно на действующих фермах и комплексах без существенных изменений технологических линий удаления навоза.
2.4.1. Жидкий навоз должен быть предварительно освобожден от посторонних включений, иметь влажность 90 — 96%, соотношение C:N — 10 — 18:1, зольность не более 20% (недостаток азота ограничивает процесс метанового брожения).
2.4.2. Хранение исходного навоза перед сбраживанием не должно превышать 24 — 48 ч.
2.4.3. Навоз от фермы поступает в навозоприемник, оборудованный насосом с измельчающим и перемешивающим устройством, обеспечивающим гомогенизацию массы для подогревателя (специальная емкость — выдерживатель, секция микробиологического реактора). Емкости навозоприемников должны обеспечивать накопление не менее 2-суточного объема с фермы.
2.4.4. В подогревателе навоз доводят до необходимой температуры сбраживания, перемешивают и порциями подают в метантенк. Объем подогревателя должен соответствовать суточному выходу навоза с фермы.
2.4.5. Микробиологический процесс анаэробного брожения проходит по одному и тому же принципу для всех видов навоза и всех типов конструкций метантенков. Для протекания процесса анаэробной ферментации количество летучих жирных кислот в сбраживаемой массе должно быть в пределах 600 — 2000 мг/л. Питательные вещества с новыми порциями жидкого навоза должны поступать в метантенк ежесуточно.
2.4.6. Процесс метаногенеза происходит при температуре обрабатываемой массы 16 — 60 °C. Выбор температурного режима анаэробного брожения органических отходов диктуется требованиями качества конечных продуктов, т.е. степенью очистки жидкого навоза, обеззараживания, дегельминтизации, количеством метана в биогазе, климатическими и экономическими факторами.
2.4.7. Вместимость микробиологического реактора зависит от суточного объема получаемого навоза, выбранного температурного режима, суточной дозы загрузки, длительности сбраживания и степени разложения органического вещества.
2.4.8. Механические, гидравлические и воздушные (биогазом) системы перемешивания сбраживаемой массы в биореакторе обеспечивают одинаковую (единую) температуру обрабатываемого субстрата во всем объеме метантенка, разрушение поверхностных коркообразований и щадящий режим брожения. Процесс анаэробного сбраживания в метантенке ведется при избыточном давлении до 200 — 400 мм водного столба (0,2 — 0,4 кПа).
2.4.9. Количество метантенков должно быть не менее двух, обеспечивающих оптимальные условия анаэробной ферментации и позволяющих при вспышке инфекционных болезней перевести работу биореакторов с проточного на цикличный режим работы.
2.4.10. Учитывая возможность поступления необработанного навоза в зоны выпуска сброженной массы, в существующих проточных технологиях с эксплуатацией двух метантенков следует предусматривать выдерживание сброженного навоза на очистных сооружениях не менее 3 сут. в отстойниках или емкостях. При наличии трех и более метантенков для ферментации, работающих в последовательном режиме, шестисуточное карантинирование обрабатываемой массы обеспечивается и дополнительных емкостей для сброженного навоза не требуется.
В случае возникновения инфекционных болезней анаэробное сбраживание жидкого навоза осуществляют при термофильном режиме (53 — 56 °C) с выдерживанием навоза в метантенках не менее 3 сут. без добавления свежих порций необработанной массы.
При попадании контаминированного сброженного навоза в накопители обеззараживание достигается при выдерживании сброженной массы в открытом навозохранилище в течение 6 мес.
2.4.11. Внесение в метантенк микробной «закваски» из культур термофилов при оптимальном режиме термофильного сбраживания позволяет сократить сроки обеззараживания от аспорогенной микрофлоры до 1 сут.:
- температура процесса — 52 — 54 °C,- влажность обрабатываемой массы — 92 — 96%,
- концентрация гидроксильных ионов, pH, — 7,0 — 8,0,
- количество термофилов — 0,6 — 1,0 млн./мл,
- доза суточной загрузки — 10 — 20%,
- частота загрузки — 1 раз в сут.,
- количество перемешиваний массы в ферментере — 3 раза в сут.,
- продолжительность каждого перемешивания — 15 — 20 мин.,
- давление в ферментере — 0,2 — 0,4 кПа.
При термофильном режиме и равномерной температуре во всех слоях обрабатываемой массы гибель возбудителей паразитарных болезней обеспечивается в течение суток.
2.5. Из биологических методов обеззараживания жидкого навоза эффективен и метод аэробной стабилизации (интенсивного окисления) при нагревании массы до 60 °C и экспозиции в течение 4 сут. При этом достигается и дезодорация жидкого навоза.
Внесение инокулята из термофильных микроорганизмов в количестве 1 млн/г обрабатываемой массы позволяет сократить сроки обеззараживания до 2 сут.
2.6. Для реализации химического способа обеззараживания жидкого навоза свинокомплексов в состав сооружений по его подготовке к использованию должны дополнительно предусматриваться специальные емкости, насосы для перекачки и периодической гомогенизации.
2.6.1. При обеззараживании жидкого навоза формалином объем емкости для различных типоразмеров предприятий следует рассчитывать, исходя из условий дезинфекции органических отходов, только в теплый период года. Формалин вводят в количестве 0,3% (по ДВ) к обрабатываемому навозу, массу перемешивают в течение 6 ч и выдерживают 72 ч. Обеззараженный навоз может быть направлен на разделительные установки и использоваться на сельскохозяйственных угодьях с учетом требований дезинвазии его, так как формалин не обеспечивает гибели в навозе возбудителей гельминтозов.
2.6.2. Обеззараживание жидкого навоза от возбудителей инфекционных и инвазионных болезней безводным аммиаком можно проводить в любое время года, так как при его введении температура обрабатываемой массы поднимается до 20 — 25 °C. Аммиак перевозится в автоцистернах МЖА-6, ЗБА-3,2 под давлением в сосудах 6 атм., подается в навоз через специальные дозаторы или по трубе, заканчивающейся перфорированной иглой (конструкции НИПТИЖ), опускаемой на дно емкости с обрабатываемой массой. Укол иглой производят на расстоянии 1 — 2 м от стен емкости и друг от друга. Во время введения происходит перемешивание массы. Обработанный навоз покрывают эмульсионно-дезинфицирующими пленками (лизол санитарный марки «Дезонол», масляный альдегид и др.). Аммиак вводят в количестве 2 — 3%, эмульсионно-дезинфицирующие вещества 0,1 — 0,3% к обрабатываемому субстрату и выдерживают навоз в течение 3 — 5 сут.
Обеззараженные органические отходы вывозят на поля мобильным транспортом, вносить их рекомендуется внутрипочвенным методом или под плуг.
Обработанный формалином жидкий навоз по влиянию на урожай не уступает необработанному, а обработанный безводным аммиаком увеличивает урожайность сельхозкультур на 15 — 20%.
2.6.3. На свиноводческих комплексах мощностью 54 — 216 тыс. голов, имеющих в составе очистных сооружений двухступенчатую биохимическую обработку и биологические пруды, обеспечивающих глубокую очистку стоков от органических веществ (БПК5 — 12 — 16 мг О2/л, ХПК — 40 — 100 мг/л, взвешенные вещества — 20 — 25 мг/л, растворенный кислород — 6 — 10 мг/л), по согласованию с местными органами госветнадзора и госсанэпиднадзора допускается в периоды вспышки инфекционных болезней обеззараживание очищенного стока хлорированием при остаточном хлоре не менее 1,5 мг/л после 30 мин. контакта или озонированием при остаточном озоне 0,3 — 0,5 мг/л после 60 мин. контакта с тщательным перемешиванием обрабатываемых стоков.
Дозы вводимых хлора и озона подбираются в каждом конкретном случае. Учитывая тот факт, что озон легко и быстро разлагается до кислорода, снимается проблема токсичности его остатков. Озон всегда можно получить при наличии кислорода и электричества, поэтому отсутствует необходимость его хранения. Этот способ обработки стоков весьма перспективен, но требуется конкретная разработка технологий обеззараживания на различных типах озонаторов.
Сырые осадки из отстойников и избыточный активный ил могут быть обеззаражены безводным аммиаком или анаэробной ферментацией в биоэнергетических установках.
2.7. Обеззараживание неочищенных навозных стоков достигается при обработке их гамма-излучением Со-60 от вегетативной патогенной микрофлоры дозами — 2 — 12 кГр, возбудителей туберкулеза — 13 кГр, спорообразующих возбудителей — 20 кГр.
После очистки органических отходов до параметров: по взвешенным веществам — 90 — 110 мг/л, БПК5 — 115 — 130 мг/л, окисляемость — 55 мг/л — доза ионизирующего излучения, необходимая для инактивации неспорообразующей микрофлоры, снижается до 2 — 10 кГр, возбудителей туберкулеза — 11 кГр, спор микроорганизмов — 17 кГр. При обработке бесподстилочного свиного навоза и навозных стоков ионизирующим излучением (Со-60, CS-137) полная гибель яиц аскариды наступает от дозы 1,3 кГр, трихоцефала — 0,5 кГр, эзофагостом — 0,3 кГр, ооцист эймерий — 2,5 кГр. Радиорезистентность яиц гельминтов, ооцист эймерий снижается при добавлении минеральных удобрений и барботирования массы в момент облучения.
Использование для очистки стоков адсорбентов активированного угля марки АГ-3, а также активированного угля с термически обработанным осадком сточных вод (150 — 170 °C), коагулированным сернокислым аммонием (25 мг/л в соотношении 1,0 — 2,3:1) при добавлении в стоки перекиси водорода в дозе 0,6 — 0,8 мг/л при постоянном облучении адсорбционной колонки с названными адсорбентами гамма-лучами Со-60 позволяет обеззараживать очищенные стоки в потоке с мощностью дозы радиации 25 рад/с.
Выбор источника излучения определяется в каждом конкретном случае условиями проведения процесса, требуемой производительностью и эксплуатационной надежностью. Защита источников излучения должна обеспечивать отсутствие радиоактивности обрабатываемых стоков и повышение радиоактивного фона окружающей среды (НРБ-96, ОСП-87 Госатомнадзор).
2.8. Обработка прошедшего через измельчитель жидкого навоза влажностью 95 — 97% во вращающемся электромагнитном поле в аппаратах с вихревым слоем АВС-150 (индуктор которого питается переменным током напряжением 380 В и частотой 50 Гц, потребляемая мощность 1,6 кВт) с ферромагнитными частицами (d — 1 — 2 мм, l — 5 — 20 мм) в рабочей камере массой 400 — 700 г обеспечивает обеззараживание их от вегетативной патогенной микрофлоры за 60 с, а при увеличении массы ферромагнитных частиц до 800 г дезинфекция происходит за 30 с. Использование в технологической линии нескольких аппаратов АВС позволяет обеззараживать навозные стоки в потоке.
2.9. Обеззараживание стоков с помощью униполярной активации в анодной камере мембранного электролизера требует глубокой очистки их до параметров: взвешенные вещества 3 — 5 мг/л, Бпк5 — 1 — 3 мг/л, ХПК 26 — 32 мг/л, азот аммонийных солей — 1,5 — 2,0 мг/л, общая жесткость — 5,0 — 7,7 мг/л, хлориды — 270 — 300 мг/л. Дезинфекция стоков достигается за счет образования на аноде свободного активного хлора с содержанием его в стоках 17,5 — 21,5 мг/л, повышения pH раствора до 10 и более и других еще не полностью изученных факторов, при силе тока 3 — 5 А, напряжении 32 — 37 В и плотности тока на электродах — 200 А/кв. м. Время контакта стоков, прошедших анодную зону мембранного электолизера, 10 мин., смешанных катодно-анодных стоков — не менее 30 мин. с последующим выдерживанием до исчезновения хлора в сточной жидкости.
2.10. Обработка осветленных навозных стоков в аппаратах мембранной микрофильтрации на полых волокнах с диаметром пор менее 0,2 мкм под давлением жидкости 1 — 1,2 атм. сопровождается снижением сапрофитной и индикаторной микрофлоры на 97,1 — 99,4%, однако полной санации от вегетативной патогенной микрофлоры не происходит, поэтому на случай вспышки инфекционных болезней в технологической линии следует предусматривать другие химические или физические способы обеззараживания с учетом значительного снижения микрофлоры в фильтрате стоков и увеличением в тысячу раз в сгущенной фракции.
2.11. При переработке стоков свинокомплексов в рыбоводно-биологических прудах с использованием их на орошение обеззараживание от аспорогенной патогенной микрофлоры в периоды эпизоотий обеспечивается длительным (12 мес.) выдерживанием неразделенных на фракции стоков в отстойниках-накопителях или секциях навозохранилищ.
Система рыбоводно-биологических прудов обеспечивает дезинвазию очищенных сточных вод, однако необходима биотермическая обработка осадка. При этой технологии требуется периодическая (не менее 1 раза в сезон) выгрузка илового осадка из секций прудов (водорослевого и рачкового) и внесение его под запашку под сельхозкультуры, подвергаемые силосованию или термической обработке.
2.12. Обеззараживание жидкого навоза, навозных стоков, жидкой фракции и осадка из отстойников при контаминации их вегетативной и спорообразующей патогенной микрофлорой, возбудителями инвазионных болезней следует проводить термическим способом в установках со струйными аппаратами при температуре 130 °C, давлении 0,2 мПа и экспозиции не менее 10 мин. (установка конструкции ВНИИВВиМ).
2.13. Для предприятий крупного рогатого скота всех типоразмеров целесообразно применять биологический способ обеззараживания путем выдерживания навоза в секционных накопителях, в которых его и карантинируют. При использовании биологического способа обеззараживания навоза любой влажности строительства дополнительных сооружений и приобретения оборудования не требуется, так как используются секционные прифермские хранилища, предназначенные для промежуточного хранения навоза или его фракций до 6 мес. во вневегетационный период.
В случае возникновения инфекционных болезней контаминированным возбудителями навозом могут быть заняты две секции хранилища, а остальные (не менее двух) будут обеспечивать непрерывность производственного процесса. В данном случае срок хранения благополучного навоза сокращается вдвое. После окончания срока выдерживания контаминированного возбудителями инфекций навоза он используется как органическое удобрение по ранее принятой технологии.
2.14. Подстилочный навоз с влажностью до 70% обеззараживают биотермическим методом путем рыхлой укладки его в бурты с размерами: высота до 2,5 м, ширина по основанию до 3,5 м и длина произвольная.
На бетонированной площадке бурт складируют на влагопоглощающие материалы (торф, измельченная солома, опилки, обеззараженный навоз и др.) слоем 35 — 40 см и ими же укрывают боковые поверхности слоем 15 — 20 см.
При обеззараживании твердой фракции жидкого навоза биотермическим способом лимитирующие параметры для обеспечения активных процессов следующие: влажность массы до 80%, высота бурта до 3 м, ширина по основанию до 5 м.
Выделяющуюся из бурта жидкость вместе с атмосферными осадками собирают и направляют в жижесборник для дезинфекции химическим способом.
Началом срока обеззараживания подстилочного навоза и твердой фракции жидкого навоза считают день повышения температуры в средней трети бурта на глубине 1,5 — 2,5 м до 50 — 60 °C. Время выдерживания буртов в теплое время года 2 мес., в холодное — 3 мес.
Дегельминтизация твердой фракции, компоста, подстилочного навоза влажностью до 70% обеспечивается биотермическим способом, но при выдерживании в буртах в весенне-летний период не менее 1 мес., в осенне-зимний период — не менее 2 мес., а при влажности 75% — в теплый период не менее 2 мес. и в холодный — не менее 6 мес.
2.15. Подстилочный навоз крупного и мелкого рогатого скота, звероферм и птицеферм влажностью более 70% карантинируют и при вспышках инфекционных болезней обеззараживают длительным выдерживанием в секциях навозохранилищ или земляных траншеях с гидроизоляционным слоем, которые заполняют поочередно. Заполненные контаминированным вегетативными возбудителями инфекций навозом секции навозохранилищ и траншей укрывают влагопоглощающими материалами слоем 15 — 20 см и выдерживают в течение 12 мес., при контаминации навоза возбудителем туберкулеза птичьего вида — 18 мес.
Для дегельминтизации твердого свиного навоза, содержащего подстилочные материалы, накапливаемые около малых (семейных) ферм, требуется его выдерживание более года. Для ускорения обеспечения уничтожения возбудителей гельминтозов — аскаридоза, трихоцефалеза, геменолипидоза — требуется механическое перемешивание массы осенне-зимнего периода накопления и выдерживание его на площадках в течение 5 — 6 мес.
2.16. Бесподстилочный полужидкий навоз и помет с влажностью 85 — 92% можно обеззараживать путем приготовления компостов с органическими сорбентами (измельченная солома, торф, опилки, кора, лигнин) и укладкой их в бурты (п. 2.14).
Для обеспечения необходимой влажности компостируемой массы компоненты должны смешиваться в нужном соотношении с учетом содержания в них влаги.
Для приготовления компостов на основе навоза сельхозживотных влажность компонентов должна быть не более: навоза — 92%, торфа — 60%, сапропеля — 50%, отходов деревообработки — 40 — 50%, соломы — 24%.
Для приготовления компостов на основе помета кур влажность компонентов следующая: помет — 64 — 82%, торф — 50 — 60%, солома — 14 — 16%, опилки — 16 — 25%, древесная кора — 50 — 60%, лигнин — 60%, гумусные грунты — 20 — 30%, компост — 65 — 70%.
Для активного и эффективного протекания биотермических процессов в компостах должно в одинаковой мере соблюдаться каждое из следующих условий:
- оптимальная влажность компостной массы — 65 — 70%,- соотношение компонентов не менее 1:1,
- высокая гомогенность смеси,
- оптимальная реакция среды, pH, — 6,5 — 7,7,
- достаточная аэрация массы в процессе компостирования, т.е. рыхлая укладка буртов,
- положительный тепловой баланс, оптимальное соотношение C-N (углерода к азоту) 20 — 30:1.
При подъеме температуры массы до 50 — 60 °C во всех слоях бурта в течение первых 10 сут. после складирования компосты выдерживают 2 мес. в летний и 3 мес. в зимний периоды года и затем используют по принятой технологии.
Для предотвращения рассеивания возбудителей инфекционных болезней переукладка буртов не производится.
При контаминации навоза особо опасными со споровыми формами возбудителей инфекций компосты не готовят. Подстилочный навоз и осадки из отстойников сжигают. Полужидкий, жидкий навоз и стоки обеззараживают термическим способом в пароструйных установках конструкции ВНИИВВиМ.
Жижу, выделяющуюся из компостов, направляют и обеззараживают химическими дезинфицирующими средствами аналогично п. 2.6.
2.17. При ускоренном компостировании помета птицы и навоза зверей с использованием органических сорбентов (влажность массы не выше 75%) в установках различной конструкции (биореакторах) с применением систем активного вентилирования воздухом обеззараживание от вегетативной патогенной микрофлоры достигается при повышении температуры компоста до 60 — 70 °C в течение 24 — 48 ч и последующей обработке его в течение 10 — 14 сут. Внесение в компост инокулята из термофильных микроорганизмов сокращает сроки обеззараживания до 4 — 7 сут.
2.18. Технологии приготовления вермикомпостов на основе навоза сельхозживотных и помета птицы осуществляются с помощью разведения в подготовленном компосте красного калифорнийского червя и других подвидов дождевого червя (E.foetida). Субстраты для вермикомпостирования (твердая фракция навозных стоков свинокомплексов, подстилочный навоз, помет кур и др.) подготавливают путем биотермической обработки и затем используют по принятой технологии.
Вермикомпостирование проводят в цехах с набором технологического оборудования, обеспечивающим оптимальные параметры среды (температура 20 °C +/- 2,5, влажность массы компоста — не более 70%, pH — 7,0 +/- 0,5) для маточной вермикультуры. Маточную культуру вносят в компост в количестве 30 — 50 экземпляров на 1 кг субстрата, влажность поддерживают на уровне не более 70%.
Цех и площадки для вермикомпостирования располагают с подветренной стороны от производственного сектора на расстоянии не менее 60 м.
Вермикомпост (биогумус) бывает готов к употреблению через 4 — 5 мес. после закладки в субстраты культуры красного калифорнийского червя.
Биомассу червя отделяют от субстрата и используют в качестве белковой добавки в корм животным с учетом требований ГОСТ 17536-82 «Мука кормовая животного происхождения, ТУ».
Склад для приема готовой продукции (биогумус, биомасса червя) отделяют стеной от технологического оборудования цеха и в местах сообщения оборудуют дезковрики, чтобы исключить вторичное обсеменение условно-патогенной микрофлорой получаемых продуктов.
2.19. При содержании мелкого рогатого скота на решетчатых полах с накоплением бесподстилочного навоза влажностью 89 — 93% в подпольных каналах температура в нем близка к температуре окружающего воздуха и биотермические процессы там отсутствуют, поэтому в случае вспышки инфекционных болезней его необходимо обеззараживать путем длительного выдерживания в прифермских навозонакопителях или приготовлением компостов с влагопоглощающими материалами (п. 2.14).
При содержании крупного и мелкого рогатого скота на решетчатых полах с добавлением соломы и сбором подстилочного навоза в подпольных навозохранилищах температура навоза с влажностью 65 — 70% поднимается до 50 — 55 °C и индикаторная микрофлора в титрах менее 1,0 выделяется только из верхнего слоя в 50 см. Поэтому для обеззараживания такого навоза подпольных хранилищ, контаминированного вегетативной патогенной микрофлорой, необходимо после удаления животных укрыть его влагопоглощающими материалами слоем 20 — 30 см и выдерживать не менее 1 мес. летом и 2 мес. — зимой. При большей влажности навоза из хранилища удаляют жижу и обеззараживают ее химическими средствами, а оставшийся плотный навоз выдерживают 10 — 12 мес.
Дегельминтизация полужидкого навоза крупного и мелкого рогатого скота в подпольных навозохранилищах достигается выдерживанием его в течение 5 мес.
2.20. Глубокая несменяемая подстилка при выращивании молодняка крупного рогатого, мелкого рогатого скота и птицы не обеззараживается в процессе накопления, так как температура в ней не поднимается выше температуры окружающей среды и биотермические процессы отсутствуют.
В случае возникновения инфекционных болезней животных и птицы контаминированную возбудителями глубокую подстилку после рыхления верхнего слоя складируют в бурты принятых размеров для биотермической обработки на подготовленных площадках. В таких буртах активные биотермические процессы наблюдаются уже через 48 ч, но они не равномерны даже в одном слое, поэтому их также выдерживают не менее 2 мес. летом и 3 мес. зимой.
2.21. Органические удобрения, получаемые на основе переработки подстилочного навоза, твердой фракции жидкого навоза животных и помета кур с помощью копрофагов по технологиям, разработанным Новосибирским аграрным университетом, ВИЖ, НИИЭМ, остаются контаминированными условно-патогенной микрофлорой, содержащейся в перерабатываемых субстратах. Данная технология переработки органических отходов (Т — 33 °C) не обеспечивает дезинфекции и дезинвазии обрабатываемой массы, требуется дополнительная термическая обработка ее. При термосушке вторичных продуктов в режиме выше 138 °C и экспозиции 10 мин. инактивируются возбудители гельминтозов и вегетативная патогенная микрофлора.
При использовании личинок копрофагов в качестве белкового корма для животных он должен соответствовать ГОСТ 17536-82 («Мука кормовая животного происхождения, ТУ»).
2.22. Обработка помета на крупных птицефабриках высушиванием в пометосушильных установках барабанного типа с прямоточным и противоточным движением сырья и теплоносителя обеспечивает обеззараживание его от патогенных бактерий, вирусов и возбудителей гельминтозов. Обеззараживание помета в прямоточных установках достигается при температуре входящих газов 800 — 1000 °C, выходящих — 120 — 140 °C и экспозиции не менее 30 мин. В противоточных установках (УСПП-1) обеззараживание обрабатываемой массы обеспечивается при температуре входящих газов 600 — 700 °C, в барабане 220 — 240 °C и выходящих 100 — 110 °C при экспозиции 50 — 60 мин. Влажность высушенного помета не должна превышать 10 — 12%, а общее микробное обсеменение — 20 тыс. микробных клеток в 1 г.
Часть 3
Контроль обеззараживания органических удобрений
3.1. Отбор проб органических удобрений для бактериологического контроля проводят по истечении сроков экспозиции при различных способах обеззараживания, изложенных выше в соответствующих разделах.
3.2. Лабораторный контроль за эффективностью обеззараживания органических удобрений, получаемых на комплексах и фермах в периоды вспышек инфекционных болезней животных и птицы, осуществляют микробиологическими методами по выживаемости индикаторных (санитарно-показательных) микроорганизмов: бактерий группы кишечных палочек, стафилококков и спор рода Bacillus в соответствии с «Инструкцией по лабораторному контролю очистных сооружений на животноводческих комплексах», М., 1980, и «Инструкцией по проведению ветеринарной дезинфекции объектов животноводства», М., 1989.
3.3. При анаэробной ферментации жидкого навоза и помета контроль обеззараживания проводят по выживаемости кишечной палочки и энтерококков.
3.4. При контаминации органических удобрений возбудителями туберкулеза качество обеззараживания их контролируют по выживаемости стафилококков и энтерококков, так как сапрофитные микробактерии не только сохраняют жизнеспособность более длительно, чем патогенные виды, но и размножаются при длительном хранении органических отходов.
3.5. Качество обеззараживания при обсеменении органических отходов спорообразующими возбудителями сибирской язвы, эмфизематозного карбункула, брадзота, злокачественного отека, а также возбудителями экзотических инфекций контролируют по наличию или отсутствию аэробных спорообразующих микроорганизмов рода Bacillus.
3.6. Обеззараживание органических отходов считают эффективным при отсутствии в 10 г (куб. см) пробы кишечных палочек, стафилококков, энтерококков или аэробных спорообразующих микроорганизмов в зависимости от вида возбудителей инфекционных болезней при трехкратном исследовании.
3.7. Качество обеззараживания навоза, навозных стоков, твердой и жидкой фракций, илового осадка от возбудителей паразитарных болезней, в том числе социально опасных, контролируют по наличию погибших яиц аскариды, трихоцефала, эзофагостом, фасциол, личинок стронгилят, яиц крысиного цепня, цист простейших и ооцист эймерий, яиц клещей в единице объема отбираемой пробы (100 г осадков иловой фракции, 100 — 500 г твердой фракции или навоза в 1 — 10 л жидкой фракции, жидкого навоза, навозных стоков) при трехкратной повторности исследований.
3.8. Бактериологический и гельминтологический (паразитологический) контроль обеззараживания навоза, помета и стоков и на их основе компостов осуществляют специалисты ветеринарных лабораторий.
Контроль за эксплуатацией технологических линий подготовки органических удобрений осуществляют специалисты ветеринарной службы предприятий.
Ответственность за выполнение настоящих «Правил» возлагается на руководителей предприятий.
3.9. Исследования проб проводят в соответствии с методиками, изложенными в Приложениях 1, 2.
Часть 4
Хранение и транспортирование
4.1. Жидкий, полужидкий навоз и навозные стоки накапливают и хранят в специальных навозохранилищах секционного типа. Подстилочный навоз, твердую фракцию жидкого навоза и компосты обрабатывают и хранят на площадках с твердым покрытием.
4.2. Вместимость навозохранилищ рассчитывают исходя из суточного количества выхода навоза и времени его использования.
4.3. Навозохранилища, предусмотренные для хранения неразделенного на фракции навоза, должны быть оборудованы устройствами для его перемешивания. Скосы и днища навозохранилищ должны иметь твердое покрытие. В закрытых навозохранилищах должны быть предусмотрены люки и приточно-вытяжная вентиляция.
4.4. Транспортирование всех видов навоза, стоков и продуктов их переработки осуществляют передвижным транспортом или стационарными устройствами (гидромеханический транспорт).
Часть 5
Использование навоза и навозных стоков
5.1. Использование навоза, помета и животноводческих стоков в качестве органических удобрений на сельскохозяйственных угодьях должно осуществляться с учетом охраны окружающей среды от загрязнений и безопасности для здоровья людей и животных. Для этого необходимо предусматривать мероприятия, исключающие:
- загрязнение поверхностных и подземных вод,
- инфицирование животных при контакте с поливной водой, почвой и выращиваемыми культурами.
5.2. Выбор участков для использования навоза и стоков в качестве органических удобрений, экспертиза проектов оросительных систем и приемка в эксплуатацию этих объектов должны проводиться с участием представителей государственной ветеринарной службы.
5.3. При выборе участков для использования органических удобрений необходимо предусматривать наличие потребных площадей сельхозугодий с учетом систем удаления, обработки и утилизации, санитарно-защитных зон и лесных насаждений.
5.4. Навоз и животноводческие стоки должны транспортироваться, обрабатываться и использоваться отдельно от хозяйственно-бытовых, производственных и смешанных сточных вод (в том числе от жилых поселков). Допускается сброс бытовых стоков от отдельных санузлов, расположенных в животноводческих помещениях, на очистные сооружения животноводческого комплекса.
5.5. Строительство оросительных систем должно завершаться до ввода комплексов в эксплуатацию.
5.6. Использование навоза и стоков в растениеводстве необходимо осуществлять, избегая повреждений или загрязнений сельскохозяйственных культур, а также не вызывая отдаленных последствий влияния на человека и животных.
5.7. Дозы внесения азота, фосфора и калия определяются их выносом урожаем сельхозкультур с учетом коэффициентов использования.
5.8. При использовании среднеструйных и дальнеструйных дождевальных установок необходимо учитывать скорость движения ветра и его направление.
5.9. При внесении навоза и животноводческих стоков в вегетационный период должен соблюдаться срок между последним удобрительным поливом и сбором урожая или его использованием.
5.10. Искусственная биологическая очистка жидкой фракции навоза свиноводческих предприятий допускается в исключительных случаях при недостатке пригодных земельных площадей и воды для орошения, а также при неблагоприятных климатических, географических и гидрогеологических условиях и в случае передачи на городские сооружения канализации.
«Ветеринарно-санитарные правила подготовки к использованию в качестве органических удобрений навоза, помета и стоков при инфекционных и инвазионных болезнях животных и птицы» разработаны Всероссийским научно-исследовательским институтом ветеринарной санитарии, гигиены и экологии и Всероссийским институтом гельминтологии имени К.И. Скрябина.
Приложение 1
Методики подготовки проб органических удобрений и исследование их на наличие индикаторных микроорганизмов
1. После обеззараживания пробы подстилочного, твердой фракции и полужидкого навоза отбирают с разных уровней навозохранилищ или буртов по диагонали не менее 100 г из каждой точки в стерильную посуду. В лаборатории навески проб растирают в фарфоровой ступке, добавляют 1:5 — 10 стерильной водопроводной воды или физиологического раствора и фильтруют через двойной слой марли.
Пробы навозных стоков и очищенных сточных вод отбирают с помощью пробоотборников в стерильные флаконы (V — 500 мл), транспортируют и хранят по общепринятым методикам.
Для исследований фильтрат навозных проб и сточные воды центрифугируют при 3000 об./мин., центрифугат в объеме 1 мл вносят в жидкие накопительные среды с последующим пересевом из пробирок, где обнаружен рост, на плотные селективные среды.
Пробы навоза и стоков после дезинфекции химическими средствами также фильтруют, фильтрат центрифугируют, центрифугат 2 — 3 раза отмывают стерильным физиологическим раствором или водопроводной водой. Отмытый осадок ресуспендируют в 1 мл стерильного физиологического раствора или водопроводной воды, высевают в жидкие элективные среды с последующим пересевом на селективные плотные среды для индикации и идентификации выделенных микроорганизмов.
2. Для индикации кишечной палочки центрифугат засевают в пробирки с глюкозопептонной средой обычного состава и поплавками в соотношении 1:5 и инкубируют в термостате 24 ч при температуре 43 °C.
Из каждой пробирки с глюкозопептонной средой, где отмечено помутнение, образование газа и кислоты, делают посевы петлей штрихами на поверхности среды Эндо в чашках Петри, разделенных на 3 — 4 сектора. Посевной материал берут таким образом, чтобы получить изолированные колонии. Чашки с посевами помещают в термостат крышками вниз и инкубируют 18 — 20 ч при 37 °C.
Типичные колонии кишечной палочки, выросшие на среде Эндо, круглой формы, гладкие, выпуклые или с приподнятой в центре поверхностью, с ровными краями, розового, красного или малинового цвета с металлическим блеском или без него. Однако учитываются и бесцветные колонии, так как дезинфектаны могут влиять на цвет колоний.
Из двух-трех разного типа колоний каждого сектора приготовляют мазки, окрашивают по Граму и микроскопируют, а также проверяют у них оксидазную активность. Колонии грамотрицательных и оксидазонегативных бактерий засевают в полужидкую среду с глюкозой и инкубируют 4 — 5 ч при 37 °C. Сбраживание сахара с образованием кислоты и газа указывает на наличие кишечной палочки.
3. Для индикации стафилококков центрифугат в объеме 1 мл вносят в солевой МПБ с 6,5% хлорида натрия в соотношении 1:5 и инкубируют посевы в термостате в течение 24 — 48 ч при 37 °C. Из пробирок, в которых обнаружен рост бактерий, делают пересев на агар Чепмена в чашках Петри и инкубируют посевы при тех же условиях. Из характерных круглых, выпуклых и окрашенных колоний (белые, лимонного или оранжевого цвета) с агара Чепмена готовят мазки, окрашивают их по Граму и микроскопируют. Наличие в мазках грампозитивных кокков, расположенных в виде виноградных гроздьев, говорит о присутствии стафилококков.
4. Индикацию энтерококков (Str.faecalis) осуществляют посевом центрифугата в жидкую щелочно-полимиксиновую среду с последующим пересевом из пробирок, в которых обнаружен рост бактерий, на плотную глюкозо-дрожжевую среду с ТТХ. Посевы инкубируют при 37 °C по 24 — 48 ч.
С глюкозо-дрожжевой среды отсевают на МПА характерные мелкие, выпуклые с красным центром колонии для проверки биохимических свойств по тестам Шермана (рост в МПБ с pH 9,6, солевого МПБ и др.). Наличие характерных признаков указывает на присутствие энтерококков.
5. Для индикации спорообразующих аэробных микроорганизмов фильтраты проб прогревают 30 мин. на водяной бане при 65 °C, затем центрифугируют и осадок засевают в МПБ и на 2 чашки МПА. Посевы инкубируют 24 — 48 ч при 37 °C. Наличие колоний на МПА и помутнение МПБ свидетельствует о присутствии спор аэробных микроорганизмов.
Приложение 2
Питательные среды
1. Глюкозопептонная среда
Среда нормальной концентрации содержит:
- пептон — 10,0 г
- хлористый натрий — 5,0 г
- глюкозу — 5,0 г
- дистиллированную воду — 1000 мл.
После растворения указанных ингредиентов прибавляют индикатор (2 мл 1,6% спиртового раствора бромтимолового синего или 10 мл индикатора Андраде), устанавливают pH 7,4 — 7,6 и разливают среду по 10 мл в пробирки с поплавками, стерилизуют в автоклаве при 112 °C (0,5 кг/см) в течение 12 мин.
2. Среда Эндо
Готовят из сухого препарата по рецепту на этикетке.
3. Полужидкая среда с индикатором ВР и глюкозой
Готовят по рецепту на этикетке. Срок хранения — не более 7 сут.
4. Приготовление реактива для определения оксидазной активности бактерий: 30 — 40 мг а-нафтола растворяют в 2,5 мл ректифицированного этилового спирта, добавляют 7,5 мл дистиллированной воды и 40 — 60 мг диметил-п-фенилендиамина. Раствор готовят непосредственно перед определением.
5. Агар Чепмена
- МПА — 100 мл
- Хлористый натрий — 8,0 г
- Маннит — 1,0 г
- Фенольный красный — 0,0025 г.
Среду разливают в колбы и стерилизуют при 0,5 атм. в течение 20 мин.
6. Щелочно-полимиксиновая среда состоит из 3 частей:
а) дрожжевой экстракт (автолизат) — 2 мл
- глюкоза — 1,0 г
- хлористый натрий — 0,5 г
- бульон — 40 мл
б) углекислая сода — 0,53 г
- вода дистиллированная — 25 мл
в) двуосновной фосфорнокислый натрий — 0,25 г
- воды дистиллированной — 25 мл.
Все три части среды раздельно стерилизуют при 112 °C в течение 12 мин. После стерилизации смешивают, устанавливают pH 10,0 — 12,0 и добавляют полимиксин в количестве 200 ЕД/мл.
7. Глюкозопептонная среда с ТТХ и кристаллическим фиолетовым
- Дрожжевой экстракт — 2 мл
- Глюкоза — 1,0 г
- 0,01% водный раствор кристал. фиолетового — 1,25 мл
- ТТХ — 0,01 г
- 2% МПА — 100 мл
- Красители прибавляют к готовой стерильной среде перед разливкой.
Приложение 3
Виды навоза и способы его обеззараживания
| Наименование | Способы обеззараживания | ||
|---|---|---|---|
| биологические | химические | физические | |
| Подстилочный навоз влажностью 65 — 70% | биотермический | ||
| Подстилочный навоз влажностью 70 — 85% | длительное выдерживание | ||
| Твердая фракция жидкого навоза влажностью до 80% | биотермический | ||
| Навоз из подпольных хранилищ | биотермический, длительное выдерживание | ||
| Глубокая несменяемая подстилка | биотермический, длительное выдерживание | ||
| Бесподстилочный: | |||
| полужидкий с влажностью 86 — 92% | компостирование, длительное выдерживание | аммиак, формальдегид | |
| Жидкий с влажностью 93 — 97% | анаэробная термофильная ферментация, длительное выдерживание, интенсивное аэробное окисление | аммиак, формальдегид | термический, гамма-излучение, переменное электромагнитноеполе |
| Навозные стоки влажностью более 97% | длительное выдерживание | термический, гамма-излучение | |
| Биологически очищенные навозные стоки | длительное выдерживание | хлор, озон | термический, гамма-излучение |
| Осадки из отстойников | анаэробная термофильная ферментация, компостирование | аммиак, формальдегид | термический, гамма-излучение |
| Помет | компостирование, длительное выдерживание | высушивание | |
| Помет с подстилкой | биотермический, длительное выдерживание | ускоренное компостирование интенсивной вентиляцией воздухом |
Приложение 4
Максимальные сроки выживаемости возбудителей инфекционных болезней во внешней среде
| Наименование болезни | Объект внешней среды | Сроки выживаемости |
|---|---|---|
| Туберкулез | вода | 12 мес. |
| почва | 36 мес. | |
| пастбища | 24 мес. | |
| навоз | 24 мес. | |
| Бруцеллез | вода | 2,5 мес. |
| почва | 7 мес. | |
| корма | 4,5 мес. | |
| навоз | 5,5 мес. | |
| Сальмонеллез | вода | 4 мес. |
| почва | 5 мес. | |
| корма | 3 мес. | |
| навоз | 12 мес. | |
| пастбища | 11 мес. | |
| Колибактериоз | навоз | 12 мес. |
| Туляремия | вода | 6 мес. |
| почва | 2,5 мес. | |
| корма | 4,5 мес. | |
| Ку-лихорадка | вода | 5 мес. |
| навоз | 12 мес. | |
| Орнитоз | вода | 17 сут. |
| навоз | 4 мес. | |
| Листериоз | вода | 18 мес. |
| почва | 18 мес. | |
| корма | 5,5 мес. | |
| навоз | 11 мес. | |
| Дерматомикозы | почва | 18 мес. |
| навоз | 3 мес. | |
| Бешенство | вода | 36 мес. |
| Ящур | вода | 20 сут. |
| почва | 10 мес. | |
| корма | 7 мес. | |
| пастбища | 1 мес. | |
| навоз | 5,5 мес. | |
| Болезнь Ауэски | корм, вода, опилки, навоз, осенне-зимний период | 19 — 60 сут. |
| лето | 7 — 20 сут. | |
| почва и трава | 12 ч — 5 сут. | |
| Лептоспироз | речная, прудовая, озерная вода | до 10 сут. |
| колодезная вода | 10 — 12 мес. | |
| навозная жижа | до 8 — 24 ч | |
| влажная почва | до 6 мес. | |
| Рожа свиней | жидкий навоз | 6 — 6,5 мес. |
| Некробактериоз | моча | 15 сут. |
| фекалии | до 2 мес. | |
| Везикулярная болезнь свиней | навоз, контаминированныепомещения | не менее 2 мес. |
| Пастереллез | помет | 2,5 мес. |
| Болезнь Марека | помет | 6 мес. |
| Болезнь Гамборо | на поверхностях внутри помещения | до 4 мес. |
| вода, корма, помет | 2 мес. | |
| Оспа | помет | 6 мес. |
| Инфекционный бронхит птиц | поверхности внутри помещения | 4 — 21 сут. |
| поверхности вне помещения | до 2 мес. | |
| вода вне помещения зимой | до 4 мес. | |
| вода внутри помещения | до 15 сут. | |
| Гепатит утят | влажный помет | 21 — 37 сут. |
| Атипичная чума (болезнь Ньюкасла) | помет | 1 мес. |
| Кокцидиоз | помет | 12 мес. |
| Скачать базу одним архивом |
| Скачать обновления |
| История создания базы |
| Карта сайта |
Скачать Инструкция по лабораторному контролю очистных сооружений на животноводческих комплексах
Дата актуализации: 10.08.2017


Инструкция по лабораторному контролю очистных сооружений на животноводческих комплексах
| Статус: | не действует |
| Название рус.: | Инструкция по лабораторному контролю очистных сооружений на животноводческих комплексах |
| Дата добавления в базу: | 01.09.2013 |
| Дата актуализации: | 05.05.2017 |
| Область применения: | Инструкция включает правила организации лабораторий по контролю за работой очистных сооружений, методики отбора и хранения проб жидкого навоза и продуктов его переработки, проведение химических, санитарно-бактериологических и гельминтологических анализов, методы оценки результатов контрольных измерений, правила техники безопасности при работе в лаборатории и предназначена для использования в лабораториях действующих и строящихся комплексов по производству животноводческой продукции. |
| Оглавление: | Часть I Часть II Часть III |
| Разработан: | ЛИСИ Минвуза РСФСР ВИГИС ГипроНИсельхоз (Институт Гипронисельхоз ) НИПТИМЭСХ Росгипрониисельстрой Свинопром РСФСР ВНИИВС |
| Утверждён: | 17.11.1980 Министерство сельского хозяйства СССР (USSR Ministry of Agriculture ) |
| Принят: | Минздрав СССР (USSR Minzdrav ) Главсельстройпроект Минсельхоза СССР (Glavselstroyproject, USSR Minselkhoz ) Главживпром СССР (USSR Glavzhivprom ) |
| Расположен в: | Техническая документация Экология СЕЛЬСКОЕ ХОЗЯЙСТВО Земледелие и лесоводство Животноводство и селекция животных |
| Нормативные ссылки: |
|

 «Часть I. Организация лаборатории. Методы санитарно-бактериологического и гельминтологического анализа сточных вод»
«Часть I. Организация лаборатории. Методы санитарно-бактериологического и гельминтологического анализа сточных вод»Скачать
ИНСТРУКЦИЯ
ПО ЛАБОРАТОРНОМУ КОНТРОЛЮ ОЧИСТНЫХ СООРУЖЕНИЙ
НА ЖИВОТНОВОДЧЕСКИХ КОМПЛЕКСАХ
Часть III
Определение биогенных веществ. Анализ осадков и ила.
В разработке инструкции приняли участие научно-исследовательские институты и организации: Гипронисельхоз (Н.Г.Ковалев, М.М.Еселевич, Н.С.Максимовский, И.К.Глазков, С.Д.Дурдыбаев, С.А.Кабанкова); Свинопром РСФСР (Ю.М.Ревякин); совхоз «50 лет СССР» Калининской обл. (П.П.Смирнов); Укрниигипросельхоз (О.П.Смирнов), НИИТИМЭСХ Нечерноземной зоны РСФСР (В.Н.Афанасьев), ЛИСИ (Б.Г.Мишуков), ВНИИМЖ (Ю.Е.Шуть, В.И.Нетеча), ВНИИВС (И.Д.Гришаев), Росгипрониисельстрой (И.П.Можайцев); ВИГИС (А.А.Черепанов).
Инструкция включает правила организации лабораторий по контролю за работой очистных сооружений, методики отбора и хранения проб жидкого навоза и продуктов его переработки, проведение химических, санитарно-бактериологических и гельминтологических анализов, методы оценки результатов контрольных измерений, правила техники безопасности при работе в лаборатории и предназначена для использования в лабораториях действующих и строящихся комплексов по производству животноводческой продукции.
Инструкция согласована с Главным управлением ветеринарии Министерства сельского хозяйства СССР, Главживпромом СССР, Главсельстройпроектом, Минздравом СССР.
УТВЕРЖДЕНА Министерством сельского хозяйства СССР 17 ноября 1980 г.
1. АЗОТ. ФОРМЫ АЗОТА
В сточных водах животноводческих комплексов содержатся различные соединения азота — аммонийный азот, азот органических веществ, нитритов и нитратов. При составлении балансовой схемы очистки загрязнений сточных вод (биохимическая очистка) учитывают содержание и соотношение разных соединений азота.
Сточные воды комплексов содержат азотистые вещества мочи и твердых экскрементов — мочевину, мочевую кислоту, аммиак и прочие соединения (креатин, креатинин, пуриновые основания, гиппуровую кислоту, эфиросерные кислоты, пигменты мочи и гормоны).
Мочевина животных подвергается гидролизу уробактериями с образованием аммонийных солей
Аммиачный азот переходит в окисленные формы азота — нитриты и нитраты. Процесс нитрификации — это окисление азота аммонийных солей в нитриты и нитраты. Он проходит в две стадии.
1. 2NH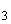 +3O
+3O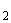 =2HNO+2HO+158 ккал
=2HNO+2HO+158 ккал
Под действием бактерий рода Nitrosomonas аммиак переходит в азотистую кислоту:
2. 2HNO+O=2HNO+38 ккал.
Под действием бактерий рода Nitrobacter происходит окисление азотистой кислоты в азотную. На окисление 1 мг аммонийного азота затрачивается 3,4 мг кислорода.
Определять формы азота в сточных водах необходимо для характеристики их биологической ценности. Соединения азота и фосфора включают в группу биогенных соединений, так как они связаны с жизнедеятельностью микроорганизмов, находящихся в воде. При значительном содержании их в водоеме наблюдается массовое развитие водорослей (цветение).
Нитриты и нитраты могут служить источником кислорода в анаэробных условиях. Восстановление бактериями солей азотной кислоты называется денитрификацией
2HNO N+HO+2,5O
N+HO+2,5O
При денитрификации на 1 мг азота приходится 2,86 мг выделившегося кислорода.
1.1. Азот аммонийный
1.1.1. Колориметрическое определение аммонийного азота реактивом Несслера.
Сущность метода. Метод основан на реакции взаимодействия иона аммония с реактивом Несслера, в результате чего образуется йодистый меркураммоний желтого цвета [4, 7]
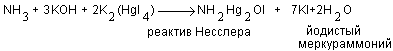
Качественное определение. К 10 мл пробы прибавляют несколько кристалликов или капель сегнетовой соли и 0,5 мл реактива Несслера.
Желтое окрашивание раствора, помутнение при выпадении желто-коричневого осадка указывает на присутствие аммиака. При содержании повышенного количества органических веществ, особенно гуминовых кислот (вызывают усиление коричневой окраски после подщелачивания), необходимо провести холостой опыт, добавив сегнетову соль и 0,5 мл 15%-ного раствора едкого натра.
Количественное определение. Подготовка пробы. Определению могут мешать взвешенные вещества и мутность воды. Если мутность воды после фильтрования не устраняется и сточные воды содержат коллоидные вещества, необходимо пробу осветлить. Для этого к 100 мл пробы прибавляют 1 мл 12,5%-ного раствора алюмокалиевых квасцов и раствор аммиака до получения рН 5,8. После взбалтывания колбы дают осадку осесть до полного осветления пробы. Фильтруют через бумажный фильтр (синяя лента).
Осветление можно проводить и взбалтыванием 100 мл пробы с 2 мл суспензии гидроокиси алюминия (приготовление суспензии см. Нитриты). При цветности более 30° проводят обесцвечивание активированным углем.
Определению аммония могут мешать амины, хлорамины, альдегиды, спирты и некоторые другие соединения, реагирующие с реактивом Несслера, тогда определение его проводят с отгонкой.
При жесткости воды более 2° (содержится кальций, магний, железо, сульфиды, хлор) содержание железа более 0,5 мг/л устраняют прибавлением раствора сегнетовой соли. Большое количество железа, сульфидов и муть удаляют при осветлении воды цинковой солью. К 100 мл пробы прибавляют 1 мл сульфата цинка (100 г ZnSO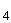 ·7HО ч.д.а, растворяют в дистиллированной воде и разбавляют до 1 л), смесь тщательно перемешивают и доводят рН до 10,5 путем добавления едкого калия или едкого натра. После взбалтывания и образования хлопьев осадок отделяют центрифугированием или фильтрованием через стеклянный фильтр. Увеличение объема жидкости необходимо учесть при расчете.
·7HО ч.д.а, растворяют в дистиллированной воде и разбавляют до 1 л), смесь тщательно перемешивают и доводят рН до 10,5 путем добавления едкого калия или едкого натра. После взбалтывания и образования хлопьев осадок отделяют центрифугированием или фильтрованием через стеклянный фильтр. Увеличение объема жидкости необходимо учесть при расчете.
Действие хлора устраняют добавлением раствора тиосульфата или арсенита натрия (растворяют в дистиллированной воде 3,5 г NaSО·5НО ч.д.а. или 1,0 г NaAsO ч.д.а. и доводят до 1 л). Для удаления 0,5 мг хлора достаточно прибавить 1 мл одного из указанных растворов.
Реактивы. 1. Безаммиачная вода. К дистиллированной воде прибавляют соду (х.ч.) до слабощелочной реакции и упаривают раствор на 1/4 объема. Проверяют воду на содержание аммиака и хранят в бутыли с тубусом. В пробку бутыли вставляют хлоркальциевую трубку, заполненную кристаллами кислого сернокислого натрия.
2. Сегнегова соль. 250 г сегнетовой соли (виннокислый калий-натрий) растворяют при нагревании в безаммиачной воде и при охлаждении объем доводят до 1 л. Добавляют 25 мл реактива Несслера и дают раствору отстояться в течение суток.
3. Реактив Несслера. Используют готовый (стандартный) реактив или приготавливают следующим образом: 7,5 г йодистого калия растирают в ступке с 3 мл дистиллированной воды, добавляют 10,5 г двуйодистой ртути и растирают до растворения, затем прибавляют 300 мл 15%-ного раствора едкого калия, перемешивают и дают отстояться в течение суток, после чего сливают прозрачный раствор, который хранят в склянке темного цвета, закрытой каучуковой пробкой. Реактив должен быть светло-желтым, прозрачным и выдерживать испытание на чувствительность к аммиаку. В цилиндр (на 100 мл) с притертой пробкой наливают 100 мл безаммиачной воды и 2 мл приготовленного реактива Несслера. В другой цилиндр (на 100 мл) наливают 100 мл безаммиачной воды, добавляют 0,005 мг аммонийного азота из рабочего раствора хлористого аммония и 2 мл реактива Несслера. Окраска во втором цилиндре должна быть интенсивнее, чем в первом.
4. Хлористый аммоний. Основной раствор — 3,819 г хлористого аммония, предварительно высушенного при 90 °С, растворяют в дистиллированной воде (безаммиачной) и доводят объем до 1 л. 1 мл этого раствора содержит 1 мг аммонийного азота.
Рабочий раствор хлористого аммония — 10 мл основного раствора доводят до объема 1 л, добавляя безаммиачную воду, 1 мл раствора содержит 0,01 мг аммонийного азота.
Калибровочная кривая. В мерные колбы на 100 мл, наполовину наполненные безаммиачной водой, добавляют 1, 2, 3, 4, 5… 50 мл рабочего раствора хлористого аммония, 0,5 мл сегнетовой соли и 1 мл реактива Несслера. Заполняют оставшийся объем колбы до метки безаммиачной водой. Через 15 мин измеряют оптическую плотность на фотоколориметре с синим светофильтром и строят калибровочную кривую по общепринятым методикам [4, 5, 6, 7].
Ход определения. Прямое определение аммиака проводят тогда, когда сточная вода после фильтрования прозрачная и бесцветная. Если вода мутная и окрашена, то аммиак определяют после предварительного осветления. В мерную колбу на 100 мл, наполовину наполненную безаммиачной водой, вносят фильтрат, содержащий 0,005-0,06 мг аммиачного азота, если измерения оптической плотности проводят в кювете с толщиной водного слоя 0,5 см. Если толщина водного слоя 1 см, то вносят 0,025-0,03 мг аммиачного азота, прибавляют 0,5 мл сегнетовой соли и 1 мл реактива Несслера, разбавляют безаммиачной водой до объема 100 мл. Через 15 мин определяют оптическую плотность окрашенного желтого раствора на фотоколориметре с синим светофильтром. Когда в пробах появляется муть, то добавляют больше сегнетовой соли и реактива Несслера при постоянном перемешивании. Отрицательного влияния на результат опыта сегнетова соль не оказывает.
Из результата оптической плотности пробы вычитают результат оптической плотности холостого опыта (безаммиачная вода и все прибавленные реактивы).
Содержание аммонийного азота в пробе определяют по калибровочной кривой.
Расчет. Количество аммонийного азота (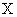 , мг/л) с учетом разбавления пробы вычисляют по формуле
, мг/л) с учетом разбавления пробы вычисляют по формуле
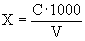 ,
,
где 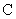 — концентрация аммонийного азота, определенная по калибровочной кривой, мг;
— концентрация аммонийного азота, определенная по калибровочной кривой, мг;
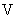 — объем пробы, взятой на определение, мл;
— объем пробы, взятой на определение, мл;
1000 — единица перевода аммонийного азота, мл в л.
Для перевода азота в ион аммония умножают полученный результат на коэффициент 1,288.
1.1.2. Определение аммонийного азота с отгонкой (в присутствии соединений, реагирующих с реактивом Несслера.
Сущность метода. Определение основано на выделении аммиака перегонкой из щелочной среды и определении его в дистилляте объемным методом с реактивом Несслера [5, 7]. Пробу подщелачивают фосфатным буферным раствором до получения [рН 7,4.]
В зависимости от содержания в пробе аммиака для его поглощения в приемнике применяют серную кислоту разной нормальности. Количество серной кислоты, израсходованной на нейтрализацию перегнанного аммиака, определяют обратным титрованием раствором едкого натра. Титруют по метиловому красному индикатору или со смешанным индикатором. Метод применяют при большом количестве аммиака в пробе. Прибавлением тиосульфата или арсенита натрия к раствору устраняют влияние хлора. Кальций в концентрациях, превышающих 250 мг/л, оказывает влияние на установление рН. В этом случае раствор подщелачивают, добавляя 40 мл буферного раствора, и смесь обрабатывают кислотой или щелочью до рН 7,4.
При отгоне аммиака определению мешает сероводород, так как при рН 7,4 около 35% сероводорода находится в свободном состоянии и он будет испаряться вместе с аммиаком. Сероводород перед отгоном осаждают солями кадмия.
Реактивы. 1. Безаммиачная вода.
2. Буферный раствор, рН — 7,4. 14,3 г однозамещенного фосфата калия безводного (ч.д.а.) и 68,8 г фосфата калия двузамещенного безводного (ч.д.а.) растворяют в дистиллированной воде и доводят до объема 1 л. При необходимости добавляют едкий калий или соляную кислоту, поддерживают рН 7,4.
3. Серная кислота 0,05 н., 0,1 н.
4. Едкий натр 0,05 н., 0,1 н.
5. Смешанный индикатор: 2 объема 0,2%-ного раствора метилового красного индикатора, растворенного в 96%-ном спирте, смешивают с 1 объемом 0,2%-ного раствора метиленовой сини в 96%-ном спирте.
Ход определения. В перегонную колбу помешают 100 мл (или большее количество в зависимости от ожидаемого содержания аммиака) сточной жидкости, которую при необходимости разбавляют примерно до 250 мл безаммиачной водой. Сточные воды, содержащие большое количество взвешенных веществ, фильтруют. Если необходимо, сточную жидкость нейтрализуют едким натром до получения рН 7 (определяется индикаторной бумажкой).
К сточной воде прибавляют 100 мл фосфатного буферного раствора. Значение рН смеси в колбе не должно изменяться более чем на ±0,2 от рН 7,4.
В приемник наливают 25 мл серной кислоты (0,05 н.) и добавляют несколько капель смешанного индикатора. Конец холодильника погружают в эту кислоту. В приемник отгоняют 100-200 мл жидкости. По окончании отгонки (проба из отгона на аммонийный азот с реактивом Несслера) избыток серной кислоты титруют (0,05 н.) раствором едкого натра.
В отдельной колбе титруют тем же едким натром 25 мл серной кислоты (0,05 н.).
Расчет. Содержание аммонийного азота (, мг/л) определяют по формуле
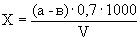 ,
,
где 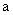 — объем раствора едкого натра (0,05 н.), израсходованного на титрование 25 мл (0,05 н.) серной кислоты, мл;
— объем раствора едкого натра (0,05 н.), израсходованного на титрование 25 мл (0,05 н.) серной кислоты, мл;
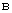 — объем раствора едкого натра (0,05 н.), израсходованного на титрование отгона, мл;
— объем раствора едкого натра (0,05 н.), израсходованного на титрование отгона, мл;
— объем анализируемой сточной воды, мл;
0,7 — количество азота, эквивалентное 1 мл (0,05 н.) раствора серной кислоты, мг;
1000 — единица перевода азота, мл в л.
1.2. Нитриты
Нитриты — промежуточный продукт бактериального окисления аммиака или восстановления нитратов. Наличие значительного количества нитритов в биологически очищенных сточных водах указывает на незавершенность процесса окисления аммонийного азота, а переход нитратов в нитриты — на недостаток кислорода, что является неблагоприятным признаком при биохимической очистке сточных вод. Вследствие химической нестойкости нитритов их следует определять сразу после отбора пробы.
1.2.1. Колориметрический метод определения нитритов с реактивом Грисса. Сущность метода. Метод основан на диазотировании сульфаниловой кислоты присутствующими в пробе нитритами и реакции получения соли с альфанафтиламином с образованием красно-фиолетового или розового азоткрасителя. Интенсивность окраски пропорциональна концентрации нитритов. Реакция протекает в кислой среде.
NО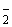 +SOНС
+SOНС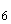 НNH+2Н
НNH+2Н SOHCНN+2HО
SOHCНN+2HО
SOНСНN+C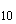 H
H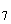 NHSOHCНN=NCHNH
NHSOHCНN=NCHNH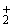 +H
+H
Ход определения. К 10 мл пробы, помещенной в пробирку, прибавляют 1 мл реактива Грисса (или 50 мг сухой навески этого реактива), перемешивают раствор и помещают на 10 мин в водяную баню (50-60 °С) или выдерживают при комнатной температуре 20-25 мин.
При содержании нитритов 0,01 мг/л появляется слабо-розовое окрашивание (при рассматривании сверху), при 0,05 мг/л — светло-розовое, при 0,1 мг/л — розовое, при 0,2 мг/л — интенсивно-розовое, при 0,5 мг/л — красное, при 1,0 мг/л — ярко-красное окрашивание.
Содержание азота нитритов можно определить и по стандартной шкале с индикатором метиловым красным. Для этого берут 10 пробирок и в каждую наливают указанное в таблице 1 количество раствора метилового красного индикатора и доводят общий объем раствора до 10 мл 10%-ной уксусной кислотой. Пробирки запаивают или плотно закрывают. Раствор устойчив в течение трех месяцев. Окраску проб сравнивают с окраской стандартной шкалы и определяют содержание нитритов в мг/л. Индикатор метиловый красный (10 мг реактива) растворяют в 1 л едкого натра (0,1 н.).
Таблица 1. Стандартная шкала для определения нитритов
|
Номера пробирок |
Объем метилового красного индикатора, прилитый в пробирку, мл |
Окраска, соответствующая содержанию азота нитритов, мл/л |
|
1 |
0,1 |
0,005 |
|
2 |
0,15 |
0,01 |
|
3 |
0,2 |
0,015 |
|
4 |
0,25 |
0,02 |
|
5 |
0,3 |
0,025 |
|
6 |
0,35 |
0,03 |
|
7 |
0,4 |
0,035 |
|
8 |
0,45 |
0,04 |
|
9 |
0,5 |
0,045 |
|
10 |
0,55 |
0,05 |
1.2.2. Количественное определение нитритов с реактивом Грисса. Подготовка проб для анализа. Определению нитритов мешают взвешенные вещества и мутность воды. Перед анализом пробу необходимо профильтровать. Если мутность после фильтрования не устраняется и сточные воды содержат коллоидные вещества, необходимо пробу осветлить коагулированием сернокислым алюминием. Для этого к 100 мл пробы прибавляют несколько капель сернокислого алюминия, доводят до рН 5,6 и после взбалтывания раствор фильтруют через бумажный фильтр (синяя лента). Осветление можно проводить также взбалтыванием 100 мл пробы с 2 мл суспензии гидроокиси алюминия.
Трехвалентное железо мешает определению нитритов, так как выпадает в осадок. Влияние его устраняют соответствующим разбавлением.
Двухвалентная медь влияет на результаты опыта вследствие вызываемого ею каталитического распада диазатированной сульфаниловой кислоты. При наличии меди пробу разбавляют дистиллированной водой, не содержащей нитриты. Определению мешает цветность воды. Слабое окрашивание и незначительную мутность воды при определении оптической плотности компенсируют холостым опытом, в котором к пробе прибавляют только раствор сульфаниловой кислоты.
Реактивы. 1. Уксусная кислота 12%-ной концентрации — 116,5 мл ледяной уксусной кислоты доводят до объема 1 л дистиллированной водой.
2. Сульфаниловая кислота. Раствор А — 0,5 г сульфаниловой кислоты растворяют в 150 мл 12%-ной уксусной кислоты.
3. 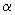 -нафтиламин. Раствор Б — 0,25 г -нафтиламина кипятят с 20 мл дистиллированной воды и отфильтровывают от нерастворившегося осадка через хорошо промытый горячей дистиллированной водой фильтр в колбу со 150 мл 12%-ной уксусной кислоты.
-нафтиламин. Раствор Б — 0,25 г -нафтиламина кипятят с 20 мл дистиллированной воды и отфильтровывают от нерастворившегося осадка через хорошо промытый горячей дистиллированной водой фильтр в колбу со 150 мл 12%-ной уксусной кислоты.
4. Реактив Грисса. 50 мл раствора А и 50 мл раствора Б перед определением опыта смешивают. Хранят раствор в склянке из темного стекла.
5. Сернокислый алюминий. 48,67 г сернокислого алюминия растворяют в дистиллированной воде и объем доводят до 250 мл.
6. Гидроокись алюминия, суспензия. Растворяют 125 г алюмокалиевых квасцов в 1 л воды, нагревают раствор до 60 °С и осторожно, при непрерывном помешивании, прибавляют 55 мл концентрированного раствора аммиака. Дают смеси постоять около 1 ч, переносят в большую бутыль (около 8 л) и промывают осадок гидроокиси алюминия дистиллированной водой до удаления хлоридов, нитритов, аммиака.
7. Дистиллированная вода (очищенная от нитритов). К 1 л дистиллированной воды приливают 1 мл концентрированной серной кислоты, 0,2 мл 50%-ного раствора сернокислого марганца и 1-3 мл 0,04%-ного раствора марганцовокислого калия. Жидкость должна окраситься в розовый цвет. Через 15 мин ее обесцвечивают, прибавляя по каплям 0,09%-ный раствор оксалата аммония.
8. Нитрит натрия. Основной раствор. Растворяют в дистиллированной воде 0,4926 г нитрита натрия (ч.д.а.), высушенного при 105 °С, и доводят объем раствора до 1 л. Раствор консервируют, добавляя 1 мл хлороформа, и сохраняют в холодном месте, он устойчив в течение месяца. 1 мл этого раствора содержит 0,1 мг азота нитритов.
Рабочий раствор N 1. Разбавляют 100 мл основного раствора дистиллированной водой до объема 1 л. Раствор должен быть всегда свежеприготовленным. В 1 мл раствора содержится 0,01 мг азота нитритов.
Рабочий раствор N 2. Разбавляют 100 мл рабочего раствора (N 1) дистиллированной водой до объема 1 л. Раствор готовят всегда перед опытом. В 1 мл раствора содержится 0,001 мг азота нитритов.
Калибровочная кривая. В мерные колбы на 100 мл, наполовину наполненные дистиллированной водой, добавляют 1-10 мл рабочего раствора (N 2), что соответствует 0,001-0,01 мг азота нитритов, и 5 мл реактива Грисса. Доводят объем раствора в мерной колбе до метки дистиллированной водой. Через 30 мин определяют оптическую плотность на фотоколориметре с зеленым светофильтром и строят калибровочную кривую по общепринятым методикам.
Ход определения. Определение нитритов проводят в прозрачной и бесцветной после фильтрования сточной воде. Если сточная вода мутная и окрашена, то нитриты определяют после предварительного осветления гидроокисью алюминия.
В мерную колбу на 100 мл наливают фильтрат, содержащий от 0,003 до 0,015 мг азота нитритов в 1-10 мл стоков, нейтрализуют раствором кислоты или щелочи до рН 6,5-7,5, добавляют дистиллированную воду, не содержащую нитриты (до половины объема колбы), 5 мл реактива Грисса и доливают дистиллированную воду до требуемой отметки. Содержимое тщательно перемешивают, затем колбу ставят в термостат на 30 мин при 20 °С. Одновременно проводят холостой опыт (дистиллированная вода с реактивом Грисса). Через 30 мин определяют оптическую плотность окрашенного раствора на фотоколориметре с зеленым светофильтром. Содержание азота нитритов вычисляют по калибровочной кривой. Результаты определения выражают в мг/л.
Расчет. Количество азота нитритов (, мг/л) рассчитывают по формуле
,
где — концентрация азота нитритов, определенная по калибровочной кривой, мг;
— объем пробы, взятой для определения, мл;
1000 — единица перевода мл в л.
Для перевода азота нитритов в нитрит-ион полученный результат умножают на коэффициент 3,284.
1.3. Нитраты
1.3.1. Качественное определение. К 2 мл сточной воды, помещенной в пробирку, по каплям прибавляют 5 мл концентрированной серной кислоты и вводят незначительное количество твердого бруцина (сильный яд!). Появившееся желтое или коричневато-красное окрашивание указывает на присутствие нитратов. Определению мешают нитриты, поэтому их предварительно удаляют азидом натрия или сульфаминовой кислотой.
1.3.2. Колориметрическое определение по Грандвалю и Ляжу. Сущность метода. При добавлении раствора фенола с серной кислотой к нитратам образуется пикриновая кислота.
3HNО+CHОH(HSО)СН(ОН)(NO)+2HSO+HО,
которая после добавления аммиака дает пикрат аммония желтого цвета
СН(ОН)(NO)+NHCH (NO)·ONH
Подготовка проб для анализа. Определению нитратов мешают взвешенные вещества, мутность и цветность воды. Для осветления воды используют суспензию гидроокиси алюминия, активированный уголь.
Если определению мешают хлориды, то их осаждают сернокислым серебром. Необходимое количество сернокислого серебра определяют на основании показателей предварительного титрования сточной воды. Берут 100 мл пробы, прибавляют 0,5 мл 10%-ного раствора хромокислого калия и титруют раствором сернокислого серебра до розового окрашивания. Количество сернокислого серебра (, мл) определяют по формуле
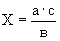 ,
,
где — количество сернокислого серебра, пошедшего на титрование, мл;
 — объем сточной воды, взятой для определения нитратов, мл;
— объем сточной воды, взятой для определения нитратов, мл;
— объем пробы, взятой на титрование, мл.
В случае почернения сухого остатка при выпаривании пробы в фарфоровой чашке (восстановление серебра) выпаривание следует повторить при меньшем количестве сернокислого серебра. При содержании в воде нитритов — более 2 мг/л — их предварительно окисляют до нитратов, для этого к объему пробы прибавляют 0,5 мл перекиси водорода и кипятят 10 мин. К значению определенного опытным путем количества нитратов вводят соответствующую поправку. При значительном содержании аммонийных солей добавляют в пробу сточной воды несколько капель 10%-ного раствора сернокислого натрия.
Реактивы. 1. Сульфофеноловый реактив. 25 г фенола (х.ч.) растворяют в 150 мл концентрированной серной кислоты (плотность 1,84) и нагревают в течение 6 ч в колбе с обратным холодильником над кипящей водяной баней. Реактив хранят в темной склянке с притертой пробкой.
2. 25%-ный водный раствор аммиака.
3. Сернокислое серебро. В 1 мл дистиллированной воды растворяют 4,44 г сернокислого серебра (1 мл раствора эквивалентен 1 мг хлорида).
4. Основной раствор нитрата калия. В дистиллированной воде растворяют 0,7216 г нитрата калия (ч.д.а.) и доводят объем раствора до 1 литра (1 мл раствора содержит 0,1 мг азота нитратов).
5. Рабочий раствор нитрата калия. 50 мл основного раствора выпаривают на водяной бане до сухого остатка. Тщательно и быстро смачивают 2 мл сульфофенолового реактива, растирают стеклянной палочкой и разбавляют дистиллированной водой до объема 500 мл (1 мл раствора содержит 0,01 мг азота нитратов).
Калибровочная кривая. В мерные колбы на 100 мл вносят 0,1; 0,3; 0,5; 0,7; 1… 30 мл рабочего раствора, что соответствует содержанию 0,001; 0,003; 0,005, 0,007, 0,01… 0,3 мг азота нитратов, добавляют 2 мл сульфофенолового реактива и такое же количество аммиака, какое прибавляют к пробе при определении нитратов. Объем раствора доводят до метки дистиллированной водой. Измеряют оптические плотности окрашенных растворов на фотоколориметре с синим светофильтром и строят калибровочную кривую по общепринятым методикам.
Ход определения. Отбирают от 10 до 100 мл осветленной сточной воды с содержанием азота нитратов в пробах от 0,01-0,05 мг и выливают в фарфоровую чашку, выпаривают на водяной бане до образования сухого остатка. В охлажденную чашку вносят 1 мл сульфофенолового реактива, тщательно растирают стеклянной палочкой, затем через 10 мин приливают 10 мл дистиллированной воды и 25%-ный раствор аммиака до получения щелочной реакции (по лакмусовой бумаге). При наличии нитратов появляется желтая окраска раствора. Если образовавшееся хлорное серебро при этом не растворится, добавляют еще несколько капель аммиака.
Жидкость переливают в мерную колбу на 100 мл и доливают до метки дистиллированную воду. Одновременно ставят холостой опыт. Определяют оптическую плотность раствора на ФЭК, применяя при этом синий светофильтр.
Расчет. Количество азота нитратов (, мг/л) вычисляют по формуле
,
где — концентрация азота нитратов, найденная по калибровочной кривой, мг;
— объем исследуемой сточной воды, мл;
1000 — единица перевода мл в л.
1.4. Общий азот. Метод Къельдаля
Сущность метода. Определение общего азота основано на разложении органических веществ сточной жидкости при нагревании с концентрированной серной кислотой, при добавлении в качестве катализатора сернокислой меди и сернокислого калия для повышения температуры реакции (температура кипения — 345-370 °С) [5, 6].
Определение азота по Къельдалю осуществляют в три стадии:
1) сжигание органических веществ серной кислотой — образуется сернокислая соль аммония;
2) отгон аммиака — нейтрализуя серную кислоту щелочью, вытесняют ион аммония из сернокислой соли с образованием свободной гидроокиси аммония, которая при нагревании разлагается до аммиака и воды:
NHОН=NH+HО
Аммиак отгоняют в приемник с серной кислотой и образуется сульфат аммония:
2NH+HSO=(NH)SO
3) косвенное определение аммиака по избытку кислоты в приемнике.
Общий азот рекомендуется определять сразу после отбора проб.
Реактивы. 1. Безаммиачная вода.
2. Серная кислота 0,1 н.
3. Едкий натр 0,1 н.
4. Концентрированная серная кислота (плотность 1,84 г/см ).
).
5. Едкий натр 50%-ной концентрации.
6. Сульфат меди 10%-ный.
7. Индикатор метиловый красный — растворяют в 7,4 мл 0,05 н. раствора едкого натра, 0,1 г метилового красного индикатора и разбавляют раствор дистиллированной водой до 100 мл,
8. Фенолфталеин, 1%-ный спиртовой раствор.
9. Сульфат калия или сульфат натрия, безводный.
10. Пемза. Кусочки пемзы кипятят несколько раз в дистиллированной воде, сливая воду после каждого кипячения, и высушивают.
Ход определения (при отсутствии в пробе нитритов и нитратов). К 10-300 мл сточной жидкости (в зависимости от количества азота), помещенной в колбу Къельдаля, прибавляют 10 мл серной кислоты (плотность 1,84 г/см), 5 г сульфата калия или сульфата натрия, 1 мл раствора сульфата меди и насыпают несколько кусочков пемзы для спокойного кипения раствора.
Содержимое колбы кипятят в вытяжном шкафу (используя специальное устройство, рис.1) до полного выпаривания воды, затем колбу закрывают втулкой и продолжают кипятить до тех пор, пока раствор в колбе не станет совсем прозрачным или слегка зеленоватого оттенка. Если нет вытяжного шкафа, используют установку с отводом сернистого газа (рис.2, 3).
Рис.1. Установка для минерализации пробы при определении общего азота
Рис.2. Установка для сжигания проб при отсутствии в лаборатории вытяжного шкафа
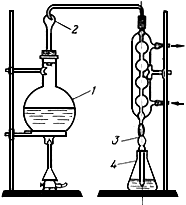
Рис.3. Прибор для отгона аммиака
Охладив колбу, переливают жидкость вместе с кусочками пемзы в колбу для отгонки аммиака (рис.4). Стенки колбы Къельдаля обмывают безаммиачной водой (250 мл). Приготовляют приемную колбу, в которую наливают 25 мл 0,1 н. серной кислоты и добавляют несколько капель раствора метилрота. Конец холодильника опускают в приемную колбу. В колбу для отгона приливают несколько капель фенолфталеина и затем по стенке колбы наливают примерно 50 мл раствора едкого натра 50%-ной концентрации. Когда жидкость окрасится в малиновый цвет, колбу быстро закрывают пробкой с каплеуловителем, перемешивают ее и доводят до кипения. Кипятят раствор в отгонной колбе до тех пор, пока не отгонят примерно половину ее содержимого. Полноту отгона аммиака проверяют качественной пробой с реактивом Несслера. Для этого конец дистилляционной трубки тщательно обмывают снаружи (над приемником), собирают две-три капли дистиллята и помещают в фарфоровую чашечку и прибавляют каплю реактива Несслера. Раствор должен остаться бесцветным или окраситься в слабо-желтый цвет.
По окончании отгонки содержимое приемной колбы титруют 0,1 н. раствором едкого натра до изменения красной окраски на оранжево-желтую.
Одновременно проводят холостое определение, величину азота, полученную при этом, вычитают из результата анализа опыта.
Расчет. Общее содержание азота (, мг/л) вычисляют по формуле
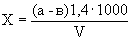 ,
,
где — объем 0,1 н. раствора едкого натра, израсходованного на титрование 25 мл серной кислоты 0,1 н., мл;
— объем 0,1 н. раствора едкого натра, израсходованного на титрование отгона, мл;
— объем анализируемой сточной воды, мл;
1,4 — количество азота, эквивалентное 1 мл 0,1 н. раствора серной кислоты, мг;
1000 — единица перевода мл в л.
Реактивы для анализа общего азота.
1. Порошкообразное железо (ч.д.а.), восстановленное.
2. Серная кислота (ч.д.а.), разбавленная (1:3).
3. Безаммиачная вода.
4. Смесь для минерализации (серная кислота+сульфат калия+сульфат ртути). Растворяют 134 г сульфата калия (ч.д.а.) в 650 мл дистиллированной воды и смешивают с 200 мл концентрированной серной кислоты (ч.д.а.). Добавляют раствор сульфата ртути (2 г окиси ртути растворяют в 25 мл 20%-ного раствора серной кислоты).
В качестве катализатора можно использовать и металлическую ртуть. После охлаждения объем смеси доводят до 1 л дистиллированной водой.
5. Раствор для подщелачивания (смесь едкого натра и тиосульфата). Растворяют 500 г едкого натра (ч.д.а.) и 25 г тиосульфата натрия в дистиллированной воде и доводят объем до 1 л.
6. Другие реактивы, необходимые для определения аммиака (см. Реактивы — общий азот).
Ход определения (при наличии в воде нитритов и нитратов). В колбу Къельдаля вносят 25-300 мл пробы (в зависимости от предполагаемого содержания общего азота), подкисляют 5 мл разбавленной серной кислотой, добавляют около 0,5 г порошкообразного железа. Содержимое колбы нагревают на водяной бане до тех пор, пока в колбе останется лишь незначительное количество железа. После охлаждения раствора добавляют 50 мл смеси для минерализации и недолго кипятят. Через 20-30 мин после осветления смеси минерализация считается законченной. Остаток смеси в колбе разбавляют безаммиачной водой, добавляют несколько капель фенолфталеина и нейтрализуют раствором для подщелачивания до слабо-розовой окраски. Колбу присоединяют к перегонному аппарату и отгоняют аммиак в приемник с кислотой (анализ и расчет см. Ход определения общего азота при отсутствии нитритов и нитратов).
Сточную воду, содержащую нитриты и нитраты, можно обработать и другим способом.
К пробе сточной воды, помешенной в колбу Къельдаля, добавляют раствор сульфата железа в таком объеме, чтобы на 1 мг азота нитратов и нитритов приходилось 80 мг этой соли. Затем приливают серную кислоту и другие реактивы и продолжают определение, как описано выше. В сильнокислой среде сульфат железа реагирует с нитратами и нитритами, в результате чего образуются окислы азота, которые улетучиваются.
Для ускорения процесса минерализации можно использовать перекись водорода, которую добавляют к сточной воде примерно через час ее кипячения с серной кислотой (при появлении белых паров сернистого газа). Перекись водорода добавляют постепенно по каплям в охлажденную пробу. После этого пробу опять нагревают до появления паров сернистого газа, охлаждают и снова приливают перекись водорода, до осветления жидкости. Остаток перекиси водорода удаляют выпариванием пробы.
Применение перекиси водорода позволяет сократить время минерализации пробы сточной жидкости до двух-четырех часов [12].
2. ФОСФАТЫ. ОБЩИЙ ФОСФОР
2.1. Колориметрическое определение ортофосфатов по Фоглеру
Сущность метода. Метод основан на реакции взаимодействия фосфатов с молибдатом аммония и серной кислотой. В качестве восстановителя фосфорно-молибденового комплекса применяют аскорбиновую кислоту в присутствии ионов трехвалентной сурьмы [5]. Чувствительность метода высокая. Минимально определяемая концентрация 0,025 мг/л. Фосфор определяют сразу после отбора пробы.
Подготовка проб для анализа. Сильнокислые и сильнощелочные пробы предварительно нейтрализуют. Определению фосфора мешают сульфиды и сероводород в концентрациях, превышающих 3 мг/л. Влияние их можно устранить прибавлением нескольких миллиграммов твердого марганцовокислого калия на 100 мл пробы и встряхиванием в течение одной-двух минут, раствор должен остаться розовым. После этого приливают раствор аскорбиновой кислоты, перемешивают и добавляют раствор молибдата аммония в серной кислоте.
Влияние нитритов устраняют сульфаминовой кислотой, которую вводят в состав применяемого реактива.
Определению мешают взвешенные вещества и мутность воды, поэтому перед анализом пробу необходимо профильтровать через плотный бумажный фильтр (синяя лента).
Если сточная вода содержит коллоидные вещества и мутность после фильтрования не устраняется, пробу пропускают через мембранный фильтр (величина пор 0,45 мкм). Для фильтрования коллоидных частиц можно применить «бумажное тесто». Берут обрезки фильтровальной бумаги и помещают их в банку с притертой пробкой. В банку наливают немного воды, закрывают пробкой и энергично взбалтывают. Бумага быстро расслаивается на волокна, образуя кашицу. Содержимое выливают в фарфоровую воронку Бюхнера и отсасывают воду насосом.
Во время отсасывания необходимо образовавшееся бумажное тесто придавливать корковой пробкой, чтобы получилась вогнутая форма для более плотного соприкосновения краев фильтра со стенками воронки. После того как бумажное тесто перенесли на воронку, его промывают (отсасывая) водой до тех пор, пока не перестанут проходить в колбу Бунзена отдельные волокна бумаги. Фильтрат получают прозрачный, фильтр («бумажное тесто») при засорении легко можно очистить.
Реактивы. 1. Смесь для анализа. 144 мл концентрированной серной кислоты ч.д.а. (плотность 1,84 г/см) приливают при помешивании к 300 мл дистиллированной воды. После охлаждения до 20 °С к кислоте при перемешивании прибавляют 10 г сульфаминовой кислоты (ч.д.а.), разведенной в 100 мл воды, 12,5 г молибдата аммония, разведенного в 200 мл воды, 0,235 г хлористой сурьмы (ч.д.а.) и 0,6 г винной кислоты (ч.д.а.) или 0,345 г антимонилтартрата калия (ч.д.а.), разведенных в 100 мл дистиллированной воды. После охлаждения до комнатной температуры объем раствора доводят до 1 л и хранят в бутыли из темного стекла.
2. Аскорбиновая кислота, 10%-ный раствор. Хранят на холоде, устойчива 30 дней.
3. Фосфат калия однозамещенный (стандартный раствор):
а) основной раствор — навеску 0,4392 г фосфата калия однозамешенного (ч.д.а.), высушенного в течение двух часов при 105 °С, растворяют в дистиллированной воде, добавляют 2 мл хлороформа и объем доводят до 1 л дистиллированной водой (1 мл раствора содержит 0,1 мг фосфора);
б) рабочий раствор готовят разбавлением основного раствора в 10 раз (1 мл рабочего раствора содержит 0,01 мг фосфора).
Калибровочная кривая. В мерные колбы емкостью 100 мл отмеряют 0,25, 0,5, 1, 2, 3, 4, 5… 50 мл рабочего раствора и доливают до метки дистиллированную воду. Определяют фосфаты, как указано ниже. Полученные величины оптических плотностей используют для построения калибровочной кривой по общепринятой методике.
Ход определения. К 100 мл (или меньшему объему, доведенному дистиллированной водой до 100 мл) пробы воды, отфильтрованной через плотный бумажный фильтр (синяя лента), прибавляют 4 мл смеси для анализа и 1 мл аскорбиновой кислоты. Смесь перемешивают. Одновременно проводят холостое определение с дистиллированной водой.
Оптическую плотность измеряют через 15 мин после перемешивания на фотоколориметре с красным светофильтром.
Учитывают оптическую плотность холостого определения.
Расчет. Количество ортофосфатов (, мг/л) вычисляют по формуле
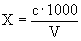 ,
,
где — концентрация фосфатов, найденная по калибровочной кривой, мг;
— объем пробы, мл;
1000 — единица перевода мл в л.
Для перевода ортофосфатов в фосфатный ангидрид полученные результаты умножают на коэффициент 2,291.
2.2. Колориметрическое определение фосфора реактивом Бартона
Сущность метода. Метод основан на реакции образования желтого растворимого фосфорно-ванадо-молибдатного комплексного соединения при взаимодействии фосфата со смешанным ванад-молибдатным реактивом в кислой среде [20].
Подготовка проб для анализа. Определению мешают хром, никель и другие ионы, имеющие собственную окраску. Мешают и восстановители. Для их окисления пробу кипятят с раствором азотной кислоты 0,5 н.
В анализируемой пробе допустимое содержание кобальта и никеля до 5 мг. Оптимально определяемое количество фосфорной кислоты при введении 5 мг ванадия — от 0,02 до 0,1 мг фосфорного ангидрида на 25 мл конечного объема пробы.
Реактивы. 1. Азотная кислота концентрированная (плотность 1,14).
2. Реактивная жидкость Бартона готовится из двух растворов.
Раствор А — 10 г молибденовокислого аммония (ч.д.а.) растворяют в 100 мл дистиллированной воды, нагретой до 50-60 °С, и при непрерывном помешивании добавляют 2 мл азотной кислоты (концентрированной). Если раствор мутный, то его фильтруют.
Раствор Б — 0,3 г ванадата аммония растворяют в 50 мл дистиллированной воды, нагретой до 50-60 °С, прибавляют 50 мл азотной кислоты, разбавленной 1:1.
Растворы А и Б смешивают, добавляют 15 мл азотной кислоты концентрированной и хранят в склянке из темного стекла. При выпадении белого творожистого осадка молибденовой кислоты раствор для определения фосфора не используют. Поэтому азотную кислоту следует приливать к неостывшему раствору молибдата аммония.
3. Перманганат калия 0,1 н.
4. Перекись водорода.
5. Фосфат калия однозамещенный (стандартный раствор).
Основной раствор — 1,9167 г фосфата калия однозамещенного (ч.д.а.), высушенного в течение двух часов при 105 °С, растворяют в дистиллированной воде, прибавляют 10 мл азотной кислоты (концентрированной) и доводят объем раствора до 1 л дистиллированной водой (1 мл раствора содержит 1 мг фосфорного ангидрида).
Рабочий раствор — разбавляют 10 мл основного раствора дистиллированной водой до объема 1 л (1 мл раствора содержит 0,01 мг фосфорного ангидрида).
Калибровочная кривая. В мерные колбы на 100 мл, наполовину наполненные дистиллированной водой, наливают 1, 2, 3, 4, 5… 50 мл рабочего раствора фосфата калия. В каждую колбу приливают 5 мл азотной кислоты (концентрированной) и 16 мл реактивной жидкости Бартона.
Одновременно ставят холостую колбу с дистиллированной водой и реактивами.
Через 15 мин после наполнения колб реактивами измеряют оптическую плотность растворов и строят калибровочную кривую по общепринятым методикам.
Ход определения. К 25 мл сточной воды (или меньшему объему, доведенному до 25 мл дистиллированной водой) приливают 10 мл азотной кислоты (концентрированной) и нагревают почти до кипения.
Органические вещества пробы разрушают при добавлении в склянку нескольких капель перманганата калия (фиолетовое окрашивание). После этого раствор снова нагревают до кипения. Избыток перманганата калия определяют по оставшемуся фиолетовому окрашиванию после кипения. Раствор слегка охлаждают и прибавляют по каплям перекись водорода для растворения перекиси марганца.
Для удаления перекиси водорода пробу выпаривают, добавляют 5 мл азотной кислоты концентрированной и 20 мл горячей дистиллированной воды. Если образуется осадок, раствор фильтруют. Прозрачный раствор переливают в мерную колбу на 100 мл, добавляют 16 мл реактивной жидкости Бартона, доводят объем раствора до требуемой нормы дистиллированной водой и тщательно перемешивают. Ставят одновременно и холостой опыт.
Через 15 мин после добавления реактивов определяют оптическую плотность окрашенных желтых растворов на фотоколориметре с синим светофильтром. Содержание фосфорного ангридида рассчитывают по калибровочной кривой.
Расчет. Количество фосфорного ангидрида (, мг/л) вычисляют по формуле
,
где — концентрация фосфора, определенная по калибровочной кривой, мг;
— объем пробы, взятый для определения, мл;
1000 — единица перевода мл в л.
Для перевода фосфорного ангидрида в общий фосфор умножают полученный результат на коэффициент 0,436.
2.3. Колориметрическое определение общего фосфора
Сущность метода. Проводят мокрое озоление сточной воды смесью хлорной и азотных кислот. Все виды фосфатов в пробе переходят в растворимые неорганические фосфаты, которые определяют молибдатом (с восстановлением аскорбиновой кислоты) в присутствии ионов трехвалентной сурьмы [7].
Подготовка проб для анализа. Определению общего фосфора мешают те же вещества, что и при определении ортофосфатов. Поэтому пробу предварительно разбавляют или применяют весовое определение.
Реактивы. 1. Смесь кислот для сжигания. Смешивают 162 мл 70%-ного раствора хлорной кислоты с 838 мл концентрированной азотной кислоты.
2. Едкий натр, раствор 10%-ной концентрации.
Остальные реактивы те же, что и при определении ортофосфатов.
Ход определения. В чашку, стакан или колбу Эрленмейера из тугоплавкого стекла отбирают 50 мл тщательно перемешанной пробы или меньшее количество, доведенное до объема 50 мл дистиллированной водой. Прибавляют 20 мл смеси кислот для сжигания и кипятят до тех пор, пока азотная кислота не улетучится и не начнут выделяться белые пары хлорной кислоты. Остаток должен составлять примерно 3 мл. После охлаждения прибавляют 5 мл дистиллированной воды и смесь снова кипятят до тех пор, пока не начнут выделяться белые пары хлорной кислоты. После повторного охлаждения к смеси прибавляют 20 мл дистиллированной воды и недолго кипятят, охлаждают, нейтрализуют раствором едкого натра и помещают в мерную колбу на 100 мл, объем доводят до метки дистиллированной водой. Продолжают определение ортофосфатов вышеприведенным способом.
Расчет такой же, как и для ортофосфатов.
2.4. Определение общего фосфора по Лоренцу
Сущность метода. Весовой метод применяют при анализе неочищенных сточных вод с большим количеством органических веществ.
Пробы консервируют, добавив 2-4 мл хлороформа на 1 л пробы. Общий фосфор определяют после минерализации сточной воды. Фосфор осаждают в виде фосфоромолибдата.
Ход определения. Отмеренное количество нефильтрованной, хорошо перемешанной пробы, содержащей не более 50 мг фосфорного ангидрида, помещают в маленькую колбочку Къельдаля и проводят озоление на слабом огне смесью серной и азотной кислот (5-10 мл) до осветления жидкости. Для определения общего фосфора также можно использовать раствор, оставшийся после определения общего азота.
Пробу охлаждают, если надо фильтруют. Общий объем раствора доводят до 50 мл, доливая дистиллированную воду. Осаждение ведут сульфатно-молибденовой жидкостью (см. Определение фосфора в осадке. Реактивы и ход определения).
3. КАЛИЙ
3.1. Определение калия кобальт-нитритным объемным методом
Сущность метода. Кобальтнитрит натрия образует с ионом калия желтый мелкокристаллический осадок
2K3Na+Co(NO) =KNa[Co (NО)]+Na
=KNa[Co (NО)]+Na
Выделение осадка происходит в нейтральной или кислой среде.
Определению калия мешают ионы аммония и органические вещества.
Калий определяют после сухого озоления сжиганием пробы в муфельной печи при темно-красном калении. Температура должна быть не более 525 °С. Пробы для определения калия не консервируют.
Реактивы. 1. Хлористый натрий — насыщенный раствор (265 г растворяют в 1 л дистиллированной воды).
2. Хлористый кобальт 10%-ной концентрации.
3. Азотистокислый натрий 10%-ной концентрации.
4. Уксусная кислота 10%-ной концентрации.
5. Сернокислый натрий 2,5%-ной концентрации.
6. Перманганат калия 0,05 н.
7. Щавелевая кислота 0,05 н.
8. Серная кислота разбавленная (1:7).
Ход определения. Пробу сточной воды, содержащую от 3 до 25 мг окиси калия, выпаривают до сухого остатка в фарфоровой чашке на водяной бане. Выпаренную пробу прокаливают в муфельной печи для удаления органических веществ и ионов аммония. Растворяют сухой остаток в 10 мл дистиллированной воды, и для осаждения калия в чашку приливают 10 мл насыщенного раствора хлористого натрия, 10 мл 10%-ного раствора хлористого кобальта и 15 мл 10%-ного раствора нитрита натрия.
Содержимое чашки тщательно перемешивают стеклянной палочкой и упаривают на водяной бане до пастообразного состояния.
Одновременно проводят холостой опыт с дистиллированной водой.
После охлаждения к пастообразному осадку приливают 10 мл 10%-ной уксусной кислоты, перемешивая содержимое чашки. Эту операцию выполняют в вытяжном шкафу, от разбрызгивания смеси чашку прикрывают стеклом и оставляют ее в вытяжном шкафу на 20-30 мин. Затем в чашку доливают 10-15 мл дистиллированной воды, перемешивают стеклянной палочной содержимое и фильтруют через стеклянный фильтр-тигель (N 4) при разрежении (вакуум). Чашку ополаскивают 2,5-ным* раствором сернокислого натрия. Осадок промывают до полного обесцвечивания и вместе с тиглем помещают в широкий химический стакан емкостью 300 мл, в который предварительно наливают 50 мл 0,05 н. перманганат калия и 10 мл разбавленной серной кислоты (1:7).
________________
* Текст соответствует оригиналу. — Примечание изготовителя базы данных.
Если тигель не залит жидкостью, то доливают дистиллированную воду.
Нагревают раствор в стакане до 70-80 °С, снимают с огня и быстро вращают тигель стеклянной палочкой. В случае обесцвечивания раствора прибавляют еще 10-20 мл перманганата калия той же нормальности. Весь процесс полного растворения осадка на фильтре должен занять не более 15-20 мин (исчезновение желтоокрашенной комплексной соли на дне стакана и фильтра). Избыток перманганата калия титруют раствором щавелевой кислоты 0,05 н. до полного обесцвечивания.
Раствором перманганата калия 0,05 н. титруют избыток щавелевой кислоты до слабого порозовения раствора, не исчезающего в течение 1 мин. 50 мл 0,05 н. перманганата калия соответствует 17,22 мг калия или 20,743 мг окиси калия.
Расчет. Содержание калия (, мг/л) вычисляют по формуле
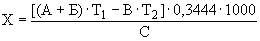 ,
,
где — объем перманганата калия, прилитого в стакан для окисления комплексной соли, мл;
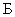 — объем перманаганата калия, израсходованного на обратное титрование, мл;
— объем перманаганата калия, израсходованного на обратное титрование, мл;
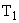 — поправка к титру перманаганата калия;
— поправка к титру перманаганата калия;
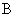 — объем щавелевой кислоты, израсходованной на титрование избытка перманаганата калия, мл;
— объем щавелевой кислоты, израсходованной на титрование избытка перманаганата калия, мл;
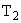 — поправка к титру щавелевой кислоты;
— поправка к титру щавелевой кислоты;
0,3444 — содержание калия (мг), в 1 мл 0,05 н. перманаганата калия (если расчет ведут на окись калия коэффициент 0,4186);
— объем сточной жидкости, взятой для выпаривания, мл;
1000 — единица перевода мл в л.
При наличии в лаборатории пламенного фотометра калий можно определить пламенно-фотометрическим методом [20]. Пламенный фотометр должен иметь фильтр для выделения спектральной линии с длиной волны 770 нм (метод описан в книге «Унифицированные методы анализа вод». — М., Химия, 1971).
Для определения специфических веществ (антибиотиков, ядохимикатов и т.д.) в обработанных сточных водах рекомендуются общепринятые методики.
4. АКТИВНЫЙ ХЛОР
Содержание активного хлора определяют в дезинфицированной сточной воде, обработанной хлором или хлорной известью.
Активный хлор — это суммарное содержание в сточной воде свободного хлора, хлорноватистой кислоты, гипохлорит-ионов и хлораминов. Определение проводят сразу после отбора пробы.
4.1. Йодометрическое определение активного хлора
Сущность метода. При подкислении анализируемой сточной воды и прибавлении к ней йодида калия все вышеперечисленные вещества выделяют йод, который титруют тиосульфатом натрия в присутствии крахмала [7].
Подготовка проб для анализа. Определению мешают окислители, выделяющие йод из йодида калия: нитриты, соли железа (трехвалентного), хлораты.
Влияние хлоратов устраняют подкислением уксусной кислотой. Если пробы сточной жидкости содержат нитриты и соли железа (трехвалентного), то титрование проводят в присутствии уксусноацетатного буферного раствора, имеющего рН 4,5.
Реактивы. 1. Тиосульфат натрия 0,01 н.
2. Йодид калия (ч.д.а.).
3. Уксусная кислота 30%-ной концентрации.
4. Крахмал 0,5%-ной концентрации.
5. Уксусноацетатный буферный раствор (рН=4,5) — смешивают 102 мл 1 н. раствора уксусной кислоты и 98 мл 1 н. раствора ацетата натрия. Объем полученного раствора доводят дистиллированной водой до 1 л. 1 н. раствор уксусной кислоты приготовляют, разбавляя 57 мл ледяной уксусной кислоты до 1 л. 1 н. раствор ацетата натрия приготовляют, растворяя 136 г ацетата натрия в воде, и доводят полученный раствор до объема 1 л.
Ход определения. В коническую колбу с притертой пробкой вливают 50-100 мл анализируемой сточной воды, вносят 0,5 г йодида калия и добавляют 10 мл уксусной кислоты. Через 5 мин оттитровывают выделившийся йод 0,01 н. раствором тиосульфата натрия (при содержании активного хлора больше 1 мг/л) или 0,005 н. раствором тиосульфата натрия (при содержании активного хлора от 0,1 до 1 мг/л). В конце титрования прибавляют 1-2 мл раствора крахмала.
Расчет. Содержание активного хлора (, мг/л) вычисляют по формуле
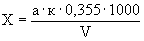 ,
,
где — объем раствора тиосульфата натрия, израсходованного на титрование пробы, мл;
 — поправочный коэффициент для приведения раствора тиосульфата натрия к 0,01 н;
— поправочный коэффициент для приведения раствора тиосульфата натрия к 0,01 н;
0,355 — количество хлора, эквивалентное 1 мл 0,01 н. раствора тиосульфата натрия, мг;
— объем анализируемой сточной воды, мл;
1000 — единица перевода мл в л.
Если анализируемая сточная вода содержит большое количество нитритов, солей железа (трехвалентного), то вместо уксусной кислоты прибавляют уксусноацетатный буферный раствор по 6 мл на каждые 100 мл анализируемой сточной воды.
В тех случаях, когда щелочность воды превышает 4 мг/экв на 1 л, количество прибавляемого буферного раствора пропорционально увеличивают (по 1,5 мл на каждый 1 мг·экв/л щелочности).
5. АНАЛИЗ ОСАДКОВ И АКТИВНОГО ИЛА
5.1. Определение влажности осадка
В фарфоровую чашку, предварительно прокаленную и взвешенную, отбирают среднюю пробу осадка 1-100 г. Осадок высушивают на водяной бане, затем в сушильном шкафу при 105 °С до постоянной массы.
Охлаждают осадок в эксикаторе и взвешивают [6].
Расчет. Влажность осадка (, %) вычисляют по формуле
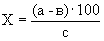 ,
,
где — масса чашки с сырым осадком, г;
— масса чашки с сухим осадком, г;
— масса осадка, г;
100 — единица перевода в %.
5.2. Определение зольности осадка
Чашку с высушенным осадком помещают в муфельную печь и прокаливают при 600 °С до постоянной массы, охлаждают в эксикаторе и взвешивают [6].
Расчет. Зольность осадка ( ) вычисляют в процентах к сухой массе осадка и определяют по формуле
) вычисляют в процентах к сухой массе осадка и определяют по формуле
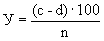 ,
,
где — масса чашки с осадком после прокаливания, г;
 — масса чашки без осадка, г;
— масса чашки без осадка, г;
 — сухая масса осадка, г;
— сухая масса осадка, г;
100 — единица перевода в %.
5.3. Определение концентрации активного ила
5.3.1. Определение концентрации активного ила по объему. Ход определения. Активный ил из аэротенков хорошо перемешивают и разливают в градуированные цилиндры на 100 мл. Через 30 мин определяют объем отстоявшегося активного ила. Результаты записывают в объемных процентах [6].
5.3.2. Определение концентрации активного ила по массе. Ход определения. Отмеренное количество иловой смеси пропускают через беззольный фильтр с белой лентой. Приставший к стенкам цилиндра ил смывают небольшим количеством дистиллированной воды. Фильтр с отфильтрованным активным илом помещают в стеклянный бюкс и высушивают в сушильном шкафу при 105 °С до постоянной массы.
Расчет. Концентрацию активного ила (, г сухой массы в 1 л) вычисляют по формуле
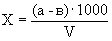 ,
,
где — масса бюкса с фильтром и илом, г;
— масса бюкса с фильтром, г;
— объем профильтрованной иловой смеси, мл;
1000 — единица перевода мл в л.
5.4. Определение зольности активного ила
Фильтр с высушенным активным илом помещают в предварительно взвешенный фарфоровый тигель, который прокаливают в муфельной печи при 600 °С до постоянной массы.
Расчет. Содержание золы (, г/л) вычисляют по формуле
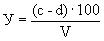 ,
,
где — масса золы и тигля, г;
— масса тигля, г;
— объем иловой смеси, взятой на определение, мл;
1000 — единица перевода мл в л.
Чаще зольность активного ила выражают в процентах к его сухой массе.
Пример расчета: концентрация активного ила — 2,86 г/л, зольность ила — 0,52 г/л. Находим содержание золы ()
2,86 — 100%;
0,52 — ;
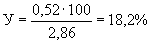 .
.
5.5. Определение илового индекса
Иловый индекс (численно) равен объему, который занимает 1 г сухого вещества активного ила через 30 мин после отстаивания в цилиндре. Пробу иловой смеси хорошо взбалтывают, наливают до черты в цилиндре (100 мл) и отстаивают в течение 30 мин. Иловый индекс (, мл/г) определяют по формуле
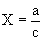 ,
,
где — объем ила, мл/л;
— сухое вещество ила, г/л.
Иловый индекс характеризует седиментационные свойства активного ила. Он изменяется в зависимости от концентрации ила в смеси, поэтому его определение ведут при дозе ила 3 г/л. Если доза ила менее 3 г/л, ил предварительно доводят до нужной концентрации отстаиванием, если больше — разбавляют водопроводной водой.
5.6. Определение общего азота по Къельдалю (в осадке и иловой жидкости)
Сущность метода и реактивы (см. Определение общего азота в сточной жидкости).
Ход определения. В стеклянный бюкс или длинную пробирку помещают 1 г средней пробы осадка (величина навески зависит от содержания азота в осадке). Навеску переносят в колбу Къельдаля. На запаянный конец пробирки надевают отрезок каучуковой трубки и с его помощью вводят пробирку в колбу Къельдаля. Пробирку опускают по возможности глубже (почти до дна колбы) и осторожно помещают навеску осадка на дно колбы. После этого пробирку снова взвешивают и устанавливают величину навески по разнице между массой пробирки с пробой и массой пустой пробирки.
К осадку в колбе прибавляют 10 мл серной кислоты (плотность 1,84), 1 мл 10%-ного раствора сульфата меди и 5 г сульфата калия. Закрывают колбу стеклянной пробкой или маленькой воронкой и кипятят в вытяжном шкафу. Нагревание начинают на слабом огне. Когда начнется сильное выделение белого дыма, нагрев увеличивают и содержимое колбы доводят до слабого кипения. Кипение серной кислоты (338 °С) все время должно быть слабым. Сильное кипение ведет к потере азота вследствие частичного разложения сульфата аммония. Одновременно проводят холостое определение на чистоту реактивов.
Сжигание органических веществ пробы кислотой считается законченным, когда раствор над минеральным остатком будет бесцветным или слабо-зеленоватым, а остаток на дне колбы — белым.
После охлаждения переносят раствор из колбы Къельдаля в перегонную колбу.
Стенки колбы Къельдаля обмывают примерно 250 мл безаммиачной водой. Прибавляют несколько капель фенолфталеина и осторожно по стенке вливают 50 мл 50%-ного раствора едкого натра. Окраска раствора в присутствии фенолфталеина должна быть ярко-розовой.
Присоединяют колбу к холодильнику, конец которого погружен в приемную колбу с 25 мл 0,1 н. серной кислоты и несколькими каплями метилрота.
Проводят отгонку аммиака. Раствор кипятят в отгонной колбе до тех пор, пока не отгонится примерно половина ее содержимого. Полноту отгонки проверяют реактивом Несслера: собирают две-три капли дистиллята в фарфоровую чашку и прибавляют каплю реактива Несслера. Раствор должен остаться бесцветным или окраситься в слабо-желтый цвет.
По окончании отгонки избыток серной кислоты в приемнике оттитровывают 0,1 н. едким натром.
Расчет. Общее содержание азота () в процентах рассчитывают по формуле
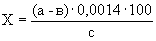 ,
,
где — количество 0,1 н. раствора едкого натра, израсходованного на титрование серной кислоты, взятой для отгонки, мл;
— количество 0,1 н. раствора едкого натра, израсходованного на титрование отгона, мл;
0,0014 — эквивалент азота, г;
— сухая навеска, г;
100 — единица пересчета в %.
5.7. Определение фосфора в осадке весовым молибденовым методом (по Лоренцу)
Сущность метода. В кислом растворе фосфорная кислота образует с молибденовокислым аммонием осадок комплексной соли фосфорно-молибденовокислого аммония. Экспериментально установлено, что в нем содержится 3,295% фосфорного ангидрида.
Кислотность раствора создают прибавлением смеси азотной и серной кислот. Жидкость перед осаждением фосфора нагревают не больше 80 °С.
В растворе, взятом для осаждения фосфора, должно содержаться фосфорного ангидрида не более 50 мг.
Полученный осадок комплексной соли фосфорно-молибденовокислого аммония промывают от примесей раствором азотнокислого аммония и ацетоном, высушивают до постоянной массы при 45-50 °С (для удаления ацетона и от пересушивания осадка комплексной соли) и по массе осадка вычисляют содержание фосфора [6, 21].
Реактивы. 1. Смесь азотной и серной кислот: 30 мл серной кислоты (плотность 1,84) приливают к 1 л азотной кислоты (плотность 1,2) и тщательно перемешивают. Азотную кислоту (плотность 1,2) получают разбавлением 424 мл концентрированной азотной кислоты (плотность 1,4) до 1 л дистиллированной воды.
2. Сульфатно-молибденовая жидкость. 100 г сульфата аммония (ч.д.а.) помещают в колбу из термостойкого стекла емкостью 2 л и растворяют при помешивании в 1 л азотной кислоты (плотность 1,36-1,37).
300 г молибдата аммония (ч.д.а.) растворяют в 700 мл горячей воды (45 °С). После охлаждения доводят объем раствора до 1 л дистиллированной водой.
Раствор молибдата аммония вливают (тонкой струей) в раствор сульфата аммония в азотной кислоте, перемешивают и оставляют стоять не менее 48 ч при комнатной температуре. После отстаивания раствор фильтруют через плотный беззольный фильтр и хранят в склянке из темного стекла в прохладном месте. Реактив сохраняет свойства в течение года,
3. Раствор азотнокислого аммония (2%-ный), подкисленный азотной кислотой до кислой среды по метилроту.
4. Ацетон, имеющий нейтральную реакцию и не содержащий альдегида. При смешивании равных объемов воды и ацетона должна быть прозрачная жидкость.
Ход определения. Для определения фосфора используют раствор, оставшийся в мерной колбе, после определения общего азота. Раствор фильтруют через плотный фильтр (синяя лента), берут 50 мл фильтрата и наливают в стакан емкостью 300-400 мл. Если указанного раствора нет, то проводят мокрое озоление — навеску средней пробы осадка 1 г (зависит от содержания фосфора) переносят в колбу Къельдаля, приливают 100 мл смеси серной и азотной кислот (1:1) и нагревают до получения прозрачного желтоватого раствора. Нагревать начинают на слабом огне.
В колбу периодически для прекращения выделения бурых паров окислов азота добавляют по 1-1,5 мл азотной кислоты (концентрированной), так как азотная кислота летуча. Если в колбе останется только серная кислота, то озоляемое вещество будет сгорать при более высокой температуре, вследствие чего могут быть потери фосфора.
Последующие порции азотной кислоты добавляют в охлажденную колбу при прекращении выделения бурых паров окислов азота из кипящего раствора в колбе.
Озоление считается законченным, когда раствор в колбе обесцветится. Охлаждают содержимое колбы, приливают немного дистиллированной воды и кипятят для удаления азотной кислоты.
После охлаждения раствор переносят в мерную колбу на 100 мл (если раствор мутный, то его фильтруют и доводят до требуемого объема дистиллированной водой).
После перемешивания из содержимого колбы отливают 50 мл раствора в стеклянный стакан емкостью 300-400 мл (остальную часть раствора можно использовать для определения калия).
К 50 мл фильтрата приливают 25 мл смеси азотной и серной кислот. Раствор нагревают до появления первых пузырьков, перемешивают, приливают 50 мл сульфатно-молибденовой жидкости. Стакан накрывают часовым стеклом и через 5 мин (не позже) перемешивают жидкость круговым вращением стакана в течение 30 сек. Выделяется желтый кристаллический осадок фосфоромолибдата аммония. Стакан с осадком оставляют стоять 12-18 ч.
Если раствор над осадком станет прозрачным через более короткий срок, то время отстаивания осадка можно сократить до трех-четырех часов.
Осадок фильтруют через высушенный и взвешенный стеклянный тигель Нутца (N 3, 4), используя для отсасывания установку с водоструйным насосом или насосом Комовского.
Осадок на фильтре промывают три-четыре раза 2%-ным раствором азотнокислого аммония, подкисленного азотной кислотой.
Проба на полноту промывания: к 10 мл фильтрата прибавляют одну каплю фенолфталеина и одну-две капли 0,02 н. едкого натра. Розовая окраска раствора показывает отсутствие кислоты в фильтрате.
Кислый раствор выливают из колбы и промывают осадок ацетоном, наполняя им тигель один раз почти до краев и два раза до половины тигля. Свежую порцию растворителя приливают тогда, когда в тигле не будет жидкости, но осадок будет влажный (не допускать высыхания). Промывание осадка органическим растворителем проводят с целью удаления остатка промывной жидкости и освобождения осадка от воды, адсорбированной поверхностью кристаллов фосфоромолибдата. Ацетон можно заменить этиловым эфиром.
Тигель вытирают снаружи и сушат 30 мин при 45 °С. Охлаждают тигель с осадком в эксикаторе без поглотителя и взвешивают.
Расчет. Содержание фосфорного ангидрида () в процентах к сухой массе осадка вычисляют по формуле
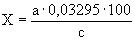 ,
,
где — масса осадка фосфорно-молибденовокислого аммония, г;
— масса средней пробы, соответствующая объему раствора, взятому на анализ, г;
0,03295 — коэффициент пересчета фосфорно-молибденовокислого аммония в фосфорный ангидрид;
100 — единица перевода в %.
Для пересчета цифрового значения фосфорного ангидрида на фосфор умножают полученный результат на коэффициент 0,436.
Для освобождения тигля от осадка промывают его несколько раз 5%-ной гидроокисью аммония и горячей дистиллированной водой. Высушивают тигель при 105 °С до постоянной массы.
5.8. Определение калия в осадке кобальт-нитритным объемным методом
(Сущность метода см. Определение калия в сточных водах)
Реактивы. 1. Уксусная кислота 10%-ный раствор — 97,1 мл ледяной уксусной кислоты разбавляют дистиллированной водой до объема 1 л.
2. Сернокислый натрий, 2,5%-ный раствор.
3. Кобальт-нитрит натрия, растворы А и Б.
А. 25 г нитрита кобальта растворяют в 50 мл дистиллированной воды и приливают 12,5 мл ледяной уксусной кислоты.
Б. 120 г нитрита натрия растворяют при осторожном нагревании в 180 мл дистиллированной воды.
За день до опыта смешивают одну часть реактива А с тремя частями реактива Б и оставляют сосуд на 1 ч открытым (часто перемешивают), затем охлаждают под струей воды в холодильнике (в течение 24 ч) и закрывают пробкой. Реактив должен быть свежеприготовленным, в холодильнике его можно хранить в течение месяца (1 мл этого раствора осаждает 3 мг калия).
Ход определения. Калий определяют после мокрого озоления осадка смесью серной и азотной кислот (см. Ход определения фосфорной кислоты весовым молибденовым методом).
Оставшуюся часть фильтра золы после определения фосфора — 50 мл (объем золы может меняться в зависимости от содержания калия), содержащих примерно 3-25 мг калия, переносят в фарфоровую чашку. Содержимое чашки выпаривают на водяной бане и прокаливают для удаления ионов аммония в муфельной печи (525 °С, темно-красное каление).
Сухой остаток в чашке после прокаливания растворяют в 3-4 мл 10%-ной уксусной кислоты и фильтруют через беззольный фильтр в фарфоровую чашку. Фильтр промывают небольшим количеством горячей дистиллированной воды, сливают промывную воду в чашку с раствором. Жидкость в чашке выпаривают на водяной бане до объема около 10 мл и прибавляют по каплям для осаждения 10 мл кобальт-нитритного реактива. Выпаривают при 80-90 °С до пастообразной консистенции, перемешивая стеклянной палочкой содержимое чашки. Охлаждают, тщательно растирают с 3 мл 10%-ной уксусной кислоты, приливают 10 мл дистиллированной воды и отфильтровывают через стеклянный фильтр (тигель) N 4 при разрежении. Осадок в тигле промывают 25%-ным раствором сернокислого натрия. Промывают до обесцвечивания вытекающего фильтрата.
Тигель с осадком переносят в стакан емкостью 500-700 мл, куда предварительно наливают 200 мл дистиллированной воды, 25-50 мл 0,1 н. перманганата калия (объем зависит от количества калия) и нагревают до 80 °С.
Горячий раствор перемешивают стеклянной палочкой две-три минуты, а затем добавляют 10 мл серной кислоты, разбавленной 1:7.
Каждые две-три минуты проводят наблюдение за растворением осадка, извлекая тигель из стакана (при помощи стеклянной палочки) или внимательно рассматривая (снизу стакана) желтый осадок, выпавший из тигля на дно стакана.
Нагревание стакана на водяной бане проводят до исчезновения желтого осадка. Цвет раствора в стакане должен быть все время фиолетовым. В случае обесцвечивания немедленно добавляют еще 5-10 мл перманганата калия 0,1 н. Количество вновь прибавленного реактива учитывают при расчете. Процесс полного растворения осадка на фильтре должен длиться не более 20 мин.
После растворения осадка (исчезновение желтой соли) в горячий раствор приливают точно отмеренное количество 0,1 н. щавелевой кислоты до полного обесцвечивания раствора.
Избыток щавелевой кислоты титруют 0,1 н. перманганатом калия до слабо-розового окрашивания.
Расчет. Вычитая из общего количества взятого перманганата калия количество щавелевой кислоты определяют содержание парманганата калия, затраченного на разрушение осадка, содержащего калий.
Содержание окиси калия в осадке (, процент сухой массы) определяют по формуле
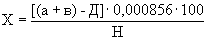 ,
,
где — объем 0,1 н. перманганата калия, прилитый в стакан для разрушения осадка комплексной соли, мл;
— объем 0,1 н. перманганата калия, пошедший на обратное титрование, мл;
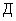 — объем 0,1 н. щавелевой кислоты, израсходованный на титрование избытка перманганата калия, мл;
— объем 0,1 н. щавелевой кислоты, израсходованный на титрование избытка перманганата калия, мл;
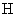 — навеска сухого осадка, соответствующая объему раствора, взятому для анализа, г;
— навеска сухого осадка, соответствующая объему раствора, взятому для анализа, г;
0,000856 — количество окиси калия, соответствующее 1 мл 0,1 н. перманганата калия, израсходованного на титрование, г;
100 — единица перевода в %.
Приложение 1
Методические рекомендации
по определению дегидрогеназной активности ила
при технологическом контроле работы аэротенков
Микроорганизмы активного ила очищают сточную жидкость от органических загрязнений за счет выделяемых ими катализаторов белковой природы и ферментов, активность которых определяет скорость и глубину процессов биологического окисления. Суммарная активность ферментов дегидрогеназ является показателем общей биологической активности ила. Дегидрогеназная активность ила обусловливается активностью самих микроорганизмов, а также количеством и степенью загрязненности сточной жидкости.
Впервые определение дегидрогеназной активности ила с ТТХ (трифенилтетразолий хлористый) предложил Букштиг. Согласно методике Букштига, для характеристики ила рекомендуется определять только так называемую исходную активность, т.е. активность ила в смеси со сточной жидкостью на данный момент очистки.
Ил характеризуется не столько изменением исходной активности, сколько изменением соотношений между активностями при добавлении к илу водопроводной воды и неочищенной сточной жидкости, т.е. субстратов. Метод применяют, если в неочищенной сточной жидкости отсутствуют токсичные вещества, а концентрация фосфатов обеспечивает процесс биологической очистки. Соотношение между величинами дегидрогеназной активности ила (исходная — 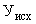 , с водопроводной водой —
, с водопроводной водой — 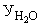 и неочищенной сточной жидкости —
и неочищенной сточной жидкости — 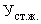 ) характеризует основные этапы процесса очистки сточных вод: для неочищенной сточной жидкости
) характеризует основные этапы процесса очистки сточных вод: для неочищенной сточной жидкости 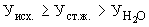 ; после изъятия загрязнений из сточной жидкости и появления нитратов —
; после изъятия загрязнений из сточной жидкости и появления нитратов — 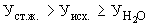 , а после регенерации активного ила —
, а после регенерации активного ила — 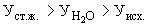 .
.
Метод позволяет установить, достигнута ли полная очистка сточных вод, произошла ли регенерация активного ила, закончился ли процесс аэробной стабилизации активного ила. Для контроля за процессом аэробной стабилизации активного ила достаточно определить 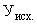 , минимальная величина которой свидетельствует об окончании процесса очистки. При этом методе анализ проводят за полтора-два часа. Не требуется сложного оборудования и специальной подготовки.
, минимальная величина которой свидетельствует об окончании процесса очистки. При этом методе анализ проводят за полтора-два часа. Не требуется сложного оборудования и специальной подготовки.
Принцип метода. Метод заключается в восстановлении бесцветной окисленной формы трифенилтетразолия хлористого (ТТХ) в красный формазан, не растворимый в воде, но растворимый в этаноле, ацетоне, бензоле и других веществах.
Количество образованного формазана (судят по интенсивности окраски) пропорционально активности дегидрогеназ.
Реактивы. 0,5%-ный раствор 2-, 3-, 5-трифенилтетразолия хлористого (ТТХ), этанол 95%-ный, соляная кислота — 0,01 н., едкий натр — 0,01 н., дехлорированная продуванием воздуха водопроводная вода, формазан (или гидросульфит натрия для получения формазана из ТТХ), глюкоза.
Варианты получения формазана.
1. 0,5 г ТТХ и 2 г глюкозы растворить в 500 мл дистиллированной воды, добавить 5 мл 2 н. щелочи (едкого кали или едкого натра), нагреть на водяной бане при 37-40 °С 10-15 мин, охладить, отфильтровать красный осадок формазана, несколько раз промыть холодной дистиллированной водой (перемешивая), просушить на воздухе в течение 48 ч, в сушильном шкафу при 30 °С 24 ч.
2. Избыток гидросульфита натрия растворяют на холоде в 20-30 мл дистиллированной воды, а 1 г ТТХ растворяют в 10 мл воды. Оба раствора сливают, а полученный осадок формазана отфильтровывают, промывают 5 раз холодной дистиллированной водой. Высушивают полученный осадок, как описано выше.
Построение калибровочной кривой. Основной раствор — 10 мг формазана разбавляют в 100 мл спирта (10 мл основного раствора содержит 1 мг формазана). Разбавляя раствор, получают соответствующие концентрации — 50, 25, 10 и 5 мкг (содержащиеся в 10 мл пробы). Измеряя оптическую плотность растворов на фотоэлектроколориметре, строят калибровочную кривую по общепринятой методике.
Аппаратура. Центрифуга (5000 об/мин). Центрифужные пробирки объемом 11 мл со стеклянными пробками. При их отсутствии используют центрифужные пробирки с резиновыми пробками и прокладками из полиэтиленовой пленки.
Отбор и подготовка проб активного ила. В связи с тем, что колебания дегидрогеназной активности ила в течение суток соответствуют колебаниям БПК поступающей сточной жидкости, отбор проб при технологическом контроле на производственных сооружениях следует проводить в одно и то же время суток. Время между отбором пробы ила и началом анализа не должно превышать 15-20 мин. Температура отобранной для анализа пробы ила должна быть доведена до 20 °С. При необходимости используют водяную баню. Температура воды в водяной бане не должна превышать 40 °С.
Ход определения. Тщательно перемешанную, нагретую до 20 °С, пробу активного ила (10-30 мг сухого вещества) по 10 мл помещают в три центрифужные пробирки (при более высокой концентрации ил разбавляется иловой водой). Добавляют по каплям 0,01 н. соляную кислоту или едкий натр во все пробирки и доводят рН до 7. Две пробирки центрифугируют 2 мин (3500-4000 об/мин), затем из них сливают надосадочную жидкость и добавляют в каждую пробирку по 10 мл водопроводной воды и неочищенной сточной жидкости после отстоя в первичных отстойниках. Содержимое этих пробирок тщательно перемешивают стеклянной палочкой, затем во все три пробирки добавляют по 1 мл 0,5%-ного раствора ТТХ. Пробирки плотно закрывают, перемешивают вручную 30 с и ставят в термостат на 55 мин при 37 °С. Одновременно в термостат ставят четвертую пробирку с илом без ТТХ (контроль).
Из термостата пробы центрифугируют 2 мин при тех же условиях. Надосадочную жидкость сливают и к осадку приливают 10 мл этанола. Содержимое пробирок перемешивают и периодически встряхивают до обесцвечивания хлопьев ила. Для полного обесцвечивания пробы центрифугируют еще 3 мин (4000-5000 об/мин). Окрашенный спиртовой раствор из каждой пробы сливают в пробирки, перемешивают и колориметрируют на ФЭК с синим светофильтром в кювете (толщина слоя 0,5 см).
Расчет. Дегидрогеназную активность выражают в мг восстановленного формазана на 1 г сухого или беззольного вещества ила (удельная активность), или на 1 л смеси ила и сточной жидкости (общая активность).
 ,
,
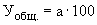 *,
*,
где — количество восстановленного формазана, мг;
— масса сухого вещества ила, г;
100* — единица перевода мл в л.
_______________
* Соответствует оригиналу. — Примечание изготовителя базы данных.
Метод обеспечивает получение точных результатов при соблюдении условий проведения анализа. Максимальное отклонение от средней величины из 10 параллельных определений составляет 3-4%.
Анализ результатов определения (пример). Анализ проведен при работе очистных сооружений с аэротенками и с отдельной регенерацией активного ила. Результаты определения дегидрогеназной активности ила, отобранного на входе и выходе из аэротенка и регенератора, приведены в табл.2.
Таблица 2. Дегидрогеназная активность ила, мг/г
|
Показатели |
Место отбора пробы |
Конец регенерации |
|
|
аэротенк |
|||
|
вход |
выход |
||
|
|
1,35 |
1,16 |
0,80 |
|
|
1,20 |
1,12 |
0,95 |
|
|
11,25 |
1,55 |
1,48 |
|
Соотношение между величинами активности ила |
|
|
|
|
Результаты опыта |
Сточная жидкость не очищена |
Изъятие загрязнений закончилось, появились нитраты, регенерация ила еще не произошла |
Регенерация ила закончилась |
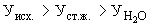
Приложение 2
Методы контроля биологической активности микроорганизмов ила
Скорость окисления сточных вод активным илом зависит от физиологического состояния микроорганизмов и определяется условиями их развития, концентрацией сточной жидкости и растворенного кислорода в очистных сооружениях и другими факторами. О физиологическом состоянии микрофлоры ила можно судить по активности ферментативных систем, которых в активном иле не менее 80-100.
Уреазная активность. Уреаза является специфическим ферментом. Она способствует гидролизу мочевины с образованием аммиака и углекислоты.
NHCONH+НО 2NH+СО
2NH+СО
Уреаза обнаружена примерно у 200 видов бактерий, большинство из которых присутствует в активном иле очистных сооружений. Определение уреазы проводится по модифицированному методу Гофмана и Шмидта. Активный ил отделяют от сточной жидкости центрифугированием и промывают два раза дистиллированной водой. Осадок ресуспензируют в дистиллированной воде так, чтобы 1 мл суспензии содержал 7-10 мг сухого вещества. К 3 мл суспензии, помещенной в коническую колбу на 100 мл с притертой пробкой, прибавляют 0,4 мл толуола и все перемешивают. Через 15 мин добавляют 3 мл цитратфосфатного буфера Мак-Ильвена (рН 7) и 3 мл 0,5%-ного водного раствора мочевины. Смесь энергично встряхивают и инкубируют при 30 °С. Через два часа реакцию останавливают добавлением 0,5 мл 10 н. соляной кислоты, смесь фильтруют через бумажный фильтр с синей лентой.
В фильтрате аммонийный азот определяют колориметрическим методом с реактивом Несслера. Количество аммонийного азота определяют по калибровочной кривой. Удельную уреазную активность рассчитывают по количеству аммонийного азота, выделенного на 1 г сухого вещества ила. В контрольные пробы вместо мочевины добавляют 3 мл воды.
Протеолитическая активность. Микрофлора очистных сооружений характеризуется высокой протеолитической активностью. Протеолитические ферменты разрывают пептидные связи в белковых молекулах, белки затем расщепляются внутриклеточными пептидазами до аминокислот, которые метаболизируются и включаются в промежуточный обмен клеток микроорганизмов.
Определение протеаз проводят по методу Хазиева и Агафаровой в модификации Архипченко и Мишукова. От сточной жидкости активный ил отделяют центрифугированием и промывают два раза дистиллированной водой. Остаток ресуспензируют в дистиллированной воде так, чтобы 1 мл суспензии содержал 7-10 мг сухого вещества. К 3 мл суспензии, помещенной в коническую колбу на 50 мл с притертой пробкой, добавляют 0,5 мл толуола и все перемешивают. Через 15 мин добавляют 5 мл 1%-ного раствора казеина в боратном буфере (рН 8,8). Смесь энергично встряхивают и инкубируют при 30 °С. Контролем служит суспензия ила без казеина.
Через 60 мин пробы центрифугируют. Переносят в пробирку 3 мл надосадочной жидкости и осаждают негидролизованный казеин, добавляя 1,5 мл раствора, содержащего 0,11 м. ТХУ, 0,22 м. ацетата натрия и 0,33 м. уксусной кислоты. Осадок отфильтровывают через бумажный фильтр и в фильтрате определяют содержание белка по методу Лоури. Допускают, что количество неосаждаемого белка пропорционально количеству тирозина, находящегося в фильтрате. Величину оптической плотности переводят по стандартной кривой (построенной по чистому тирозину) в микромоли тирозина. Удельную протеолитическую активность выражают в микромолях тирозина, выделившегося при гидролизе казеина на 1 г сухого вещества ила.
Показателем общей протеолитической активности служит аммонийный азот, выделившийся под действием микрофлоры, содержащейся в 1 л иловой смеси.
Казеин (ч.д.а.) растворяют в 1%-ном растворе углекислого натрия, подкисляют до рН 4 1%-ным раствором уксусной кислоты, отфильтровывают через марлю, промывают водой и два-три раза промывают смесью спирта и эфира, высушивают при комнатной температуре и размалывают на мельнице.
Определение белка по Лоури. Реактивы: реактив А — 2%-ный раствор углекислого натрия в 0,1 н. раствора едкого натра;
реактив В — 0,5%-ный раствор сернокислой меди в 1%-ном растворе тартрата натрия;
реактив С — готовят перед постановкой реакции смешиванием 49 частей реактива А и 1 части реактива B;
реактив Фолина — 100 г вольфрамата натрия и 25 г молибдата натрия растворяют в 700 мл дистиллированной воды. К смеси добавляют 50 мл 85%-ного раствора фосфорной кислоты и 100 мл концентрированной соляной кислоты. Смесь кипятят в колбе с обратным холодильником 10 ч, после чего в колбу добавляют 150 г сернокислого лития, 50 мл дистиллированной воды и 5 капель бромной воды. Смесь кипятят 15 мин и после охлаждения объем доводят до 1 литра. Реактив хранят в холодильнике в темной склянке;
реактив Е — готовят из реактива Фолина, разбавляя его водой (1:1) до получения 1 н. раствора.
Ход определения. К 1 мл исследуемого раствора прибавляют 2 мл реактива С и сразу интенсивно встряхивают пробу в пробирке в течение нескольких минут. Через 10 мин добавляют 0,2 мл реактива Е. После быстрого и тщательного встряхивания пробы выдерживают 40 мин в темноте и измеряют оптическую плотность на ФЭК при красном светофильтре (N 9), в кюветах с толщиной измеряемой жидкости 5 мм.
Построение калибровочной кривой. Навеску тирозина (ч.д.а.) 181,2 мг растворяют в 1 л 0,2 н. соляной кислоты, 1 мл полученного раствора содержит 1 мкм тирозина. Для построения калибровочной кривой в пробирки наливают 0,05, 0,1… 0,8 мл раствора тирозина, доливают дистиллированную воду до объема 1 мл и продолжают анализ (см. Ход определения).
Калибровочную кривую строят по общепринятой методике.
Потребление кислорода активным илом. Об окислительной способности активного ила судят по интенсивности потребления кислорода микрофлорой ила. Дыхательная активность микроорганизмов зависит от их физиологического состояния, состава и концентрации загрязнений сточной жидкости, а также от условий аэрации.
Количество кислорода, поглощенного микрофлорой, определяют при измерении парциального давления газовой фазы над суспензией активного ила в сточной жидкости. Для этого используют манометрический метод, применяя аппарат Варбурга.
Метод основан на регистрации манометром поглощенного илом кислорода при постоянной температуре. Главным условием использования этого метода для изучения газообмена является фиксация изменений давления одного газа. Парциальное давление другого газа искусственно приводится к нулю или поддерживается на определенном уровне. При изучении потребления кислорода манометром определяют кислород, а углекислый газ поглощается щелочью. Уменьшение давления, благодаря поглощению илом кислорода, вызывает падение уровня манометрической жидкости. Давление измеряется при постоянном объеме системы. Постоянство объема достигается тем, что перед отсчетом показаний манометра манометрическая жидкость в соединенном с сосудиком колене манометра всегда приводится к одной и той же отметке и принимается за нулевую. Основное уравнение манометрии
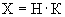 ,
,
где — количество поглощенного газа, мкл;
— изменение давления манометрической жидкости (разность между исходным уровнем манометрической жидкости в левом колене манометра и тем, который устанавливается в манометре после окончания наблюдений), мм;
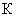 — константа сосудика — коэффициент, позволяющий перейти от давления манометрической жидкости (мл) к объему газа (мкл).
— константа сосудика — коэффициент, позволяющий перейти от давления манометрической жидкости (мл) к объему газа (мкл).
Константа сосудика показывает, какое количество поглощенного или выделявшегося газа (мкл) вызывает в данном сосудике изменение давления, равное 1 мм манометрической жидкости. При проведении экспериментальной работы возникает необходимость определения констант сосудиков. Подробное описание методов определения констант сосудиков аппарата Варбурга дано в руководствах по манометрии.
Порядок работы с аппаратом Варбурга. 1. Включить аппарат Варбурга и установить температуру воды в ванне 30 °С.
2. Проверить работу манометров и приготовить термобарометр.
3. Термобарометр — это сосудик для регистрации колебаний атмосферного давления, которое может иметь место во время опыта. Подготавливают термобарометр к работе следующим образом. Во внутренний цилиндрик сосудика вносят 0,2 мл дистиллированной воды, в сосудик — 2,0 мл дистиллированной воды. Сосудик прикрепляют к манометру и по истечении времени опыта регистрируют его показатели.
3. Подготовить к работе сосудики. Во внутренний цилиндр сосудика внести 0,2 мл 20%-ного раствора едкого калия и маленький кусочек фильтровальной бумаги (раствор едкого кали вносится для поглощения углекислого газа).
Перед проведением анализа активный ил промывают водопроводной водой. Для этого смесь ила и сточной жидкости из аэротенка помещают в пробирки и центрифугируют. Ил оседает на дно пробирки, надосадочную жидкость сливают и заменяют водопроводной водой, осадок промывают и вновь центрифугируют. После трехкратного промывания ил разводят водопроводной водой до концентрации по сухому веществу не более 2 г/л и помещают в сосудики Варбурга. Кроме водопроводной воды, можно использовать сточную жидкость (разбавленную).
Потребление кислорода илом в смеси с водопроводной водой можно принять за эндогенное дыхание ила. По количеству потребленного кислорода илом в смеси со сточной водой можно судить о скорости окисления загрязнений.
Так как расчет количества поглощенного кислорода илом ведется на 1 мг сухого вещества, то часть отмытого ила используют для определения его концентрации в сосудиках манометра.
Показания прибора через каждые полчаса записывают в таблицу. Расчет количества поглощенного кислорода проводится без данных термобарометра. По данным анализа строят графики.
Приложение 3
Методы количественного учета микроорганизмов
Микроскопический подсчет бактерий. Он дает представление об общем количестве микроорганизмов, находящихся в активном иле (живых и мертвых).
Существуют два основных метода подсчета бактерий: метод мембранных фильтров по Разумову и учет бактерий в плоскопараллельных капиллярах Перфильева.
Метод мембранных фильтров: взятую для анализа иловую смесь пропускают через мембранный фильтр, помещенный в воронку Зейца. Мембранные фильтры предварительно кипятят в дистиллированной воде, которую меняют несколько раз. Из приемного сосуда аппарата отсасывают воздух. Иловую воду профильтровывают. После фильтрования фильтр сушат на воздухе, на краях его делают карандашом пометки с указанием номера или даты профильтрованного объема воды.
Весь фильтр окрашивают карболовым эритрозином (5%-ный раствор эритрозина и 5%-ный раствор карболовой воды) в течение трех-четырех часов или оставляют в краске на ночь. Окраску проводят в чашке Петри, куда помещают кружок фильтровальной бумаги и увлажняют его краской. На смоченную краской бумагу нижней стороной кладут фильтры с осевшими взвесями и закрывают крышкой. По окончании окрашивания краску отмывают, перекладывая фильтры на листе фильтровальной бумаги, смоченной дистиллированной водой. Перекладывание фильтров с одного места на другое продолжают до тех пор, пока фильтр перестанет окрашивать влажную фильтровальную бумагу. После промывания фильтр высушивают. При этом фильтр с взвесью должен быть розового цвета, а края почти бесцветными.
Подсчет микроорганизмов проводят под микроскопом. Для этого переносят препарат на предметное стекло, наносят каплю канадского бальзама или иммерсионного масла. Накрывают покровным стеклом и исследуют под микроскопом с иммерсионным объективом (увел. х 90) при окуляре с сетчатым микрометром (увел. х 10). Подсчет бактерий () в 1 мл сточной жидкости проводят по формуле
 ,
,
где  — площадь фильтра, мм
— площадь фильтра, мм ;
;
10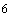 — переводный коэффициент (мм в мкм);
— переводный коэффициент (мм в мкм);
— сумма подсчитанных бактерий на участке;
— площадь окулярного сетчатого микрометра при том же увеличении, мкм;
— профильтрованная вода, мл;
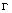 — число участков, где подсчитывались бактерии.
— число участков, где подсчитывались бактерии.
При работе с одним и тем же микроскопом и фильтровальным аппаратом величина () остается постоянной, и ее можно заменить коэффициентом (), использование которого упрощает вычисление.
Подсчет следует делать на 20 участках (поле зрения) на площади 20000 мкм в разных местах фильтра. В каждом участке должно содержаться не менее 2-5 и не более 10-20 бактерий. При большем количестве бактерий необходимо профильтровать меньший объем воды или сделать разведение водой, профильтрованной через мембранный фильтр. При получении меньших значений надо просчитать большее число участков и фильтровать большие объемы исследуемой воды.
Дезоксирибонуклеиновая кислота — показатель жизнеспособной биомассы активного ила. Скорость биохимический очистки обусловливается как физиологическим состоянием микроорганизмов, о котором можно судить по активности ферментов, так и количеством биомассы в очистных сооружениях. Определение количества биомассы осложняется тем, что в активном иле микроорганизмы окружены слизистым слоем, на котором адсорбируются инертные частицы и мелкие взвеси. Это исключает возможность определения биомассы ила высушиванием ее до постоянной массы. В то же время при эксплуатации очистных сооружений необходимо контролировать биомассу микроорганизмов без сорбированных ими инертных частиц. Особые трудности возникают при оценке активной биомассы ила в аэротенках крупных животноводческих комбинатов, так как сточные воды, поступающие на очистные сооружения, содержат большое количество непереработанных кормов, песка и других частиц. Содержание белка или общего азота не может служить показателем биомассы ила, так как эти соединения присутствуют и в виде балластных частиц в иле. Общепринятый в микробиологии метод количественного учета бактериальных клеток — подсчет колоний, выросших на мясо-пептонном агаре, нельзя рекомендовать для определения количества микроорганизмов активного ила из-за отсутствия специальных сред для смешанных культур микроорганизмов и, кроме того, из-за агрегации бактерий в хлопьях. В тех случаях, когда микроорганизмы образуют скопления, о величине биомассы можно судить по концентрации дезоксирибонуклеиновой кислоты — ДНК, так как в течение времени роста бактериальной культуры содержание ДНК в клетке практически постоянно. Этот метод в практике очистки сточных вод применяется недавно, но является перспективным.
Определение ДНК в активном иле проводят по следующей схеме. Фильтрованием через марлю отделяют грубые взвеси и полученную суспензию микроорганизмов центрифугируют. Осадок (25-30 мг сухой массы) два раза промывают физиологическим раствором, три раза раствором 0,1 м. ЭДТА + 0,15 м. поваренной соли (pH 8,2) для удаления капсульных полисахаридов и затем дважды промывают физиологическим раствором. Разделение нуклеиновых кислот ведут по методу Штидта и Таннгаузера.
Разделение нуклеиновых кислот по методу Штидта и Таннгаузера. Осадок промытых клеток ресуспензируют в физиологическом растворе с таким расчетом, чтобы 1 мл суспензии содержал 2-3 мг сухого вещества. Полученную суспензию центрифугируют (6000 об/мин 10 мин), к осадку добавляют 4 мл 1 н. едкого натра и выдерживают при 37 °С. Через два часа пробу охлаждают на ледяной бане 10 мин и доводят рН до единицы холодной концентрированной хлорной кислотой. Пробу выдерживают при 0 °С в течение 10-15 мин и далее центрифугируют (6000 об/мин 10 мин). К осадку добавляют 2 мл 1 н. хлорной кислоты. Гидролиз ДНК проводят при 80 °С в течение 20 мин и охлаждают при 0 °С (10 мин). Осадок отделяют центрифугированием, в надосадочной жидкости определяют содержание дезоксирибозы по методу Джильса и Мейерса.
Определение дезоксирибозы по методу Джильса и Мейерса. К 2 мл гидролизата ДНК в 1 н. 57%-ной хлорной кислоте добавляют 2 мл 4%-ного раствора дифениламина (ч.д.а.) в ледяной уксусной кислоте и 0,1 мл 1,6%-ного водного раствора ацетальдегида. Пробы выдерживают 18 час в пробирках с притертыми пробками при 30 °С.
В качестве стандарта используют гидролизат ДНК спермы лосося в 1 н. хлорной кислоте. Концентрацию ДНК в стандартном растворе определяют по методу Спирина на спектрофотометре СФ-16 при 270 и 290 ммк.
По данным исследований, в чистых культурах микроорганизмов концентрация ДНК составляет 1,75-5,41% от сухой биомассы клетки. В микроорганизмах активного ила очистных сооружений свинокомплексов эта величина составляет 3,5% от сухой биомассы клетки. По содержанию ДНК можно определить и количество сухой биомассы клеток ( , мг/мл) без посторонних включений, балласта и инертных частиц в активном иле по формуле
, мг/мл) без посторонних включений, балласта и инертных частиц в активном иле по формуле
 ,
,
где — концентрация ДНК, мкг/мл;
— 0,035 концентрация ДНК, в % от сухой биомассы микроорганизмов активного ила на очистных сооружениях свинокомплексов (расчетная).
Культуральные методы анализа микрофлоры активного ила. Сточные воды крупных животноводческих комплексов содержат в значительном количестве клетчатку, мочевину, белки и другие азотсодержащие соединения. Это определяет наличие в активном иле бактерий, разлагающих клетчатку, уробактерий и бактерий, участвующих в аммонификации, нитрификации, денитрификации азота.
Питательная среда для накопления нитрифицирующих бактерий.
Нитрификация протекает в две стадии. Первая — окисление аммиака до нитритов и вторая — окисление нитритов до нитратов.
Состав питательной среды для накопления бактерий для
1 фазы нитрификации
|
Сернокислый аммоний (NH |
2 г |
|
Фосфорнокислый калий, двузамещенный (K |
1 г |
|
Поваренная соль (NaCl) |
2 г |
|
Сернокислый магний (MgSO |
0,5 г |
|
Сернокислое железо (FeSO |
0,4 г |
|
Углекислый кальций (СаСО |
1 г |
|
Дистиллированная вода |
1000 мл |
Чтобы предотвратить потерю аммонийных солей, рекомендуется углекислые соли стерилизовать отдельно и после охлаждения добавлять углекислую соль в каждую колбу.
Питательную среду разливают в конические колбы слоем не более 1,5-2 см. Перед разливанием среды ее надо хорошо перемешать, так как в ней образуется взвесь фосфорнокислого железа. После стерилизации при 120 °С в течение 15 мин питательные среды засевают.
Рецепты питательных сред для количественного учета бактерий по физиологическим группам
Использование мясо-пептонного агара для учета аэробных сапрофитов. В мясо-пептонный бульон (МПБ) добавляют мелко нарезанный агар и расплавляют его. В зависимости от сорта агара добавляют его от 1,5 до 2%. После расплавления агара устанавливают рН 7,1-7,2 насыщенным раствором уксуснокислого натрия. Питательную среду разливают небольшими порциями в колбы и стерилизуют при 120 °С в течение 20 мин.
Питательная среда Гетчинсона для аэробных бактерий, разрушающих клетчатку
|
Фосфорнокислый калий двузамещенный (K |
1,0 г |
|
Хлористый кальций (СаCl |
0,1 г |
|
Сернокислый магний (MgSO |
0,3 г |
|
Хлорное железо (FeCl |
0,01 г |
|
Азотнокислый натрий (NaNO |
2,5 г |
|
Дистиллированная вода |
1000 мл |
Устанавливают рН 7,2-7,3. Питательную среду до стерилизации наливают в пробирки (1/3 объема). В каждую пробирку опускают полоску фильтровальной бумаги так, чтобы она наполовину была в растворе.
Питательная среда для уробактерий
|
Мочевина |
20 г |
|
Желатина |
150 г |
|
Мясной бульон |
1000 мл |
Устанавливают рН 7,5. Желатина может быть заменена 15 г агар-агара.
Питательная среда для аммонифицирующих бактерий
Процесс разложения азотсодержащих органических соединений с образованием аммиака называют аммонификацией. Определение количества аммонифицирующих бактерий производят методом титров, путем добавления сточной жидкости в пробирки с пептонной водой.
Состав пептонной воды:
|
Пептон |
5 г |
|
Фосфорнокислый калий, двузамещенный (K |
1,0 г |
|
Фосфорнокислый калий, однозамещенный (KH |
1,0 г |
|
Сернокислый магний (MgSO |
0,5 г |
|
Поваренная соль (NaCl) |
следы |
|
Вода водопроводная |
1 л |
Пептонную воду готовят следующим образом: в 1 л водопроводной воды вносят 5 г пептона (следует избегать избытка железа) и кипятят 15 мин, затем в горячую жидкость вносят соли. Доводят рН до 7. После растворения солей раствор тщательно взбалтывают и фильтруют до полной прозрачности. Питательную среду разливают в пробирки по 5-7 мл и стерилизуют 20 мин при 1 атм (100 кПа), ставят в термостат при 25 °С. За ростом культур ведут наблюдение начиная с четвертого дня после посева, и через сутки проводят определение на наличие азотистой кислоты реактивом Грисса в сухом виде.
Состав питательной среды для накопления бактерий во II фазе нитрификации
|
Азотистокислый натрий (NaNO |
1,0 г |
|
Углекислый натрий (Na |
1,0 г |
|
Фосфорнокислый калий двузамещенный (K |
0,5 г |
|
Хлористый натрий (NaCl) |
0,5 г |
|
Сернокислый магний (MgSO |
0,3 г |
|
Сернокислое железо (FeSO |
0,4 г |
|
Дистиллированная вода |
1000 мл |
Питательная среда Гильтая для учета денитрифицирующих бактерий
Денитрификация может идти в анаэробных и в аэробных условиях, но особенно интенсивно она протекает без доступа кислорода.
|
А. Азотнокислый калий (KNO |
1 г |
|
Аспарагин (С |
1 г |
|
Дистиллированная вода |
250 мл |
|
Б. Лимонная кислота |
5,0 г |
|
или лимоннокислый кальций |
8,5 г |
|
Фосфорнокислый калий однозамещенный (KН |
1,0 г |
|
Сернокислый магний (MgSO |
1,0 г |
|
Хлористый кальций (СаCl |
0,2 г |
|
Хлористое железо (FeCl |
следы |
|
Дистиллированная вода |
250 мл |
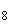 N
N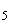 ·Н
·НВ случае употребления лимонной кислоты ее нейтрализуют 10%-ным раствором едкого кали в присутствии фенолфталеина как индикатора.
Растворы А и Б смешивают, объем доводят до 1000 мл. Наличие процесса денитрификации устанавливают по положительной реакции с реактивом Грисса и по выделению пузырей газа. Для получения твердой питательной среды добавляют 1,5% агар-агара. Среду можно употреблять и без аспарагина.
Для количественного учета денитрифицирующих бактерий берут среду Гильтая без аспарагина, добавляют к ней 1,5% агар-агара и нейтрализуют до рН 7,0-7,2. Испытуемый материал из ряда последовательных десятикратных разведений вносят в стерильные пробирки и заливают данной средой до резиновой пробки. Пробирки сразу ставят в стакан с холодной водой для быстрейшего застывания агара. О развитии бактерий судят по образованию в столбике агара пузырьков воздуха.
Приложение 4
Методы химического анализа грунтовых вод
При исследовании грунтовых вод определяют прозрачность, запах, окисляемость, нитриты, нитраты, аммонийный азот, фосфаты, хлориды.
Сроки отбора проб рекомендуется устанавливать в разные периоды года в зависимости от уровня грунтовых вод; ранней весной (до снеготаяния), в период максимального (весной) уровня грунтовых вод, к концу вегетационного периода, поздней осенью. Рекомендуется также делать отбор проб через 10 дней после внесения жидкого навоза и продуктов его переработки на поля.
В местах, подверженных наибольшему загрязнению (участки около прудов-накопителей, биологических прудов, буферных и иловых площадок и т.д.), отбор проб можно проводить чаще.
Химический анализ грунтовых вод проводят по методикам (часть II инструкции), применяемым для исследования сточных вод животноводческих комплексов, с учетом показателей, перечисленных в разделе.
Определение хлоридов в грунтовых водах
Сущность метода. Метод основан на реакции осаждения хлора азотнокислым серебром в присутствии хромовокислого калия. Пока в растворе имеются хлориды, происходит выпадение в осадок хлористого серебра, без хлора образуется хромат серебра, что вызывает появление красноватой окраски раствора
NaCl+AgNO=NaNО+AgCl
KCrO+2AgNO=AgCrО+2KNO
Этим методом можно определять хлориды в воде в пределах от 2 до 400 мг/л.
Реактивы. 1. Хлористый натрий, титрованный раствор: 1,649 г (х.ч.) хлористого натрия, высушенного при 105 °С, растворяют в мерной колбе в 1 л дистиллированной воды, 1 мл такого раствора содержит 1 мг хлора.
2. Азотнокислое серебро, титрованный раствор 4,8 г (х.ч.) нитрата серебра, растворяют в 1 л дистиллированной воды.
3. 5%-ный раствор хромовокислого калия: 50 г хромата калия растворяют в небольшом количестве дистиллированной воды, прибавляют нитрат серебра до образования легкого красного осадка, через день фильтруют и разбавляют фильтрат до объема 1 л дистиллированной водой.
Определение поправочного коэффициента нитрата серебра
В три конические колбочки наливают по 10 мл раствора хлористого натрия, доводят объем до 100 мл дистиллированной водой, прибавляют 1 мл 5%-ного раствора хромовокислого калия и титруют раствором азотнокислого серебра. После того как лимонно-желтая окраска мутного от хлористого серебра раствора перешла в оранжево-желтую окраску, не исчезающую в течение 15-20 с, титрование заканчивают.
Поправочный коэффициент к титру азотнокислого серебра (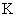 ) вычисляют по формуле
) вычисляют по формуле
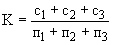 ,
,
где 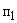 ,
, 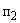 ,
, 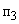 — количество азотнокислого серебра, пошедшее на каждое титрование, мл;
— количество азотнокислого серебра, пошедшее на каждое титрование, мл;
 ,
, 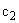 ,
, 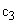 — количество раствора хлористого натрия, взятого для каждого титрования, мл.
— количество раствора хлористого натрия, взятого для каждого титрования, мл.
Ход определения. К 100 мл профильтрованной воды прибавляют 1 мл раствора хромата калия и при помешивании титруют раствором нитрата серебра до перехода лимонно-желтого окрашивания в оранжево-желтое.
Содержание хлоридов (, мг/л) в миллиграммах хлор-иона на 1 л воды вычисляют по формуле
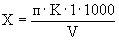 ,
,
где  — количество раствора азотнокислого серебра, пошедшего на титрование, мл;
— количество раствора азотнокислого серебра, пошедшего на титрование, мл;
— поправочный коэффициент к титру азотнокислого серебра;
 — количество хлора, эквивалентное 1 мл титрованного раствора азотнокислого серебра, мг;
— количество хлора, эквивалентное 1 мл титрованного раствора азотнокислого серебра, мг;
— объем исследуемой воды, взятой для титрования, мл;
1000 — единица перевода мл в л.
Определению хлоридов мешает сероводород, органические вещества, очень кислые или щелочные воды и большое количество железа. Кислые пробы воды нейтрализуют бикарбонатом натрия, а щелочные — азотной кислотой (по фенолфталеину) до слабо-розового окрашивания, которое устраняют затем, взбалтывая воду. Железо осаждают окисью цинка. Органические вещества разрушают кипячением 100 мл воды с кристалликом перманганата калия. Воду отфильтровывают от хлопьев перекисью марганца.
Воду, имеющую цветность выше 30 °С*, а также воду, в которой присутствует сероводород, кипятят с раствором пергидроля в течение 10 мин (1 мл 30%-ного раствора НО на 100 мл воды), после чего фильтруют.
________________
* Текст соответствует оригиналу. — Примечание изготовителя базы данных.
СПИСОК ЛИТЕРАТУРЫ
1. Положение о санитарной лаборатории на промышленном предприятии (типовое N 832-69). — М., 1969.
2. Инструкция по методическому руководству ведомственными лабораториями контроля и качества вод. — М., 1979.
3. Правила охраны поверхностных вод от загрязнения сточными водами (N 1166-74)*. — М., 1974.
__________________
* В настоящее время действующими документами в области охраны поверхностных вод являются СанПиН 2.1.5.980-00, ГН 2.1.5.1315-03, ГН 2.1.5.1316-03. — Примечание изготовителя базы данных.
4. Лурье Ю.Ю., Рыбникова А.И. Химический анализ производственных сточных вод. — М., 1974.
5. Рекомендации по методам производства анализов на сооружениях биохимической очистки промышленных сточных вод. — М., 1970.
6. Методика технологического контроля работы очистных сооружений городских канализаций. — М., 1977.
7. Унифицированные методы исследования качества воды. — М., 1975.
8. Лейте В. Определение органических загрязнений питьевых и сточных вод. — М., 1975.
9. Руководство по химическому и технологическому анализу воды. — М., 1973.
10. Коган С.Г. Биохимическая очистка сточных вод свинооткормочных комбинатов. Автореф. канд. дис. — Л., 1974.
11. Унифицированные методы анализа сточных вод. — М., 1971.
12. Гурьянова Е.М., Новикова Л.С., Бахpax И.М. Методы определения органических веществ в сточных водах от свиноферм. — В кн.: Сборник трудов ЛИСИ, 1973.
13. Преображенская А.С., Сорокина Н.И. Сравнительная оценка модифицированных методов определения растворенного кислорода и БПК в фенольных сточных водах ШПЗ в присутствии большого количества нитритов. — В кн.: Методические материалы и научные сообщения. ВНИИ железнодор. гигиены, 1971, вып. 36.
14. Общесоюзные нормы технологического проектирования систем удаления, обработки, обеззараживания, хранения и утилизации навоза и помета. — М., 1979.
15. Arial I., Humenik F. and al. Bod analysis of swine waste as affected by feed additives. Intern. simposium on livestock vastes proceeding. — USA, Columbus, Ohio 1971.
16. Евилевич И.А. О корреляции показателей БПК и ХПК при очистке сточных вод. — Химические волокна, 1975, N 3.
17. Шифрин С.М., Мишуков Б.Г., Бахpax И.М. и др. Лабораторные исследования по биохимической очистке сточных вод свинооткормочного комбината «Новый свет». — В кн.: Сборник трудов ЛИСИ. — Л., 1973.
18. Лурье Ю.Ю., Одарюк В.А. Метод определения БПК полного сточных вод с применением ингибиторов нитрификации. — Гигиена и санитария, 1975, N 5.
19. Унифицированные методы анализа вод СССР. — Л., 1978.
20. Бабко A.K., Пилипенко А.Т. Фотометрический анализ. Методы определения неметаллов. — М., 1974.
21. Петербургский А.В. Практикум по агрономической химии. — М., 1968.
22. Резников А.А., Мулиновская Е.П., Соколов И.Ю. Методы анализа природных вод. — М., 1970.
23. Научно-методическое руководство по санитарно-гельминтологическому контролю за сооружениями по переработке и хранению бесподстилочного навоза на животноводческих комплексах и фермах промышленного типа. — М., 1977.
24. Руководство по методам химического анализа морских вод. — Л., 1977.
Электронный текст документа