Содержание
-
Русское название
-
Английское название
-
Латинское название
-
Химическое название
-
Брутто формула
-
Фармакологическая группа вещества Регорафениб
-
Нозологическая классификация
-
Код CAS
-
Фармакологическое действие
-
Характеристика
-
Фармакология
-
Применение вещества Регорафениб
-
Противопоказания
-
Ограничения к применению
-
Применение при беременности и кормлении грудью
-
Побочные действия вещества Регорафениб
-
Взаимодействие
-
Передозировка
-
Способ применения и дозы
-
Меры предосторожности
-
Источники информации
-
Торговые названия с действующим веществом Регорафениб
Русское название
Регорафениб
Английское название
Regorafenib
Латинское название
Regorafenibum (род. Regorafenibi )
Химическое название
4-[4-[[4-хлоро-3-(трифторметил)фенилl]карбамоиламино]-3-фторофенокси]-N-метилпиридин-2-карбоксамид
Брутто формула
C21H15ClF4N4O3
Фармакологическая группа вещества Регорафениб
Нозологическая классификация
Список кодов МКБ-10
-
C15 Злокачественное новообразование пищевода
-
C16 Злокачественное новообразование желудка
-
C17.0 Двенадцатиперстной кишки
-
C17.1 Тощей кишки
-
C17.2 Подвздошной кишки
-
C17.3 Дивертикула Меккеля
-
C17.8 Поражение тонкого кишечника, выходящее за пределы одной и более вышеуказанных локализаций
-
C17.9 Тонкого кишечника неуточненной локализации
-
C18 Злокачественное новообразование ободочной кишки
-
C19 Злокачественное новообразование ректосигмоидного соединения
-
C20 Злокачественное новообразование прямой кишки
-
C22.0 Печеночноклеточный рак
-
C26 Злокачественное новообразование других и неточно обозначенных органов пищеварения
-
C26.9 Неточно обозначенные локализации в пределах пищеварительной системы
-
C48 Злокачественное новообразование забрюшинного пространства и брюшины
-
C48.1 Уточненных частей брюшины
-
C78.8 Вторичное злокачественное новообразование других и неуточненных органов пищеварения
Код CAS
755037-03-7
Фармакологическое действие
—
иммуномодулирующее, противоопухолевое.
Характеристика
Регорафениб является ингибитором многочисленных протеинкиназ, включая киназы, участвующие в ангиогенезе опухоли, онкогенезе, а также входящие в состав микроокружения опухоли.
RxList.com
Регорафениб представляет собой моногидрат с молекулярной массой 500,83, практически нерастворим в воде, мало растворим в ацетонитриле, метаноле, этаноле, этилацетате и умеренно — в ацетоне.
Фармакология
Фармакодинамика
Механизм действия
Регорафениб является ингибитором многочисленных протеинкиназ, включая киназы, участвующие в ангиогенезе опухоли (VEGFRl, -2, -3, TIE2), онкогенезе (KIT, RET, RAF-1, BRAF, BRAF V600E), а также входящие в состав микроокружения опухоли (PDGFR, FGFR). В частности, регорафениб ингибирует мутантную киназу KIT, ключевой онкогенный фактор в развитии стромальных опухолей ЖКТ. Благодаря этому регорафениб блокирует пролиферацию опухолевых клеток. В доклинических исследованиях было показано, что регорафениб оказывает выраженное противоопухолевое действие на широком спектре опухолевых моделей, включая колоректальный рак и гастроинтестинальные стромальные опухоли. Эффект этого ЛС связан с его антиангиогенным и антипролиферативным воздействием. Кроме того, регорафениб оказывает антиметастатическое действие in vivo. Основные метаболиты этого ЛС в организме человека (М2 и М5) по своей эффективности на моделях in vitro и in vivo сопоставимы с регорафенибом.
RxList.com (обновление 2021 г.)
Механизм действия
Регорафениб представляет собой низкомолекулярный ингибитор множества мембраносвязанных и внутриклеточных киназ, участвующих в нормальных клеточных функциях и патологических процессах, таких как онкогенез, ангиогенез опухоли, метастазирование и опухолевый иммунитет. В in vitro биохимических или клеточных анализах регорафениб или его основные активные метаболиты у человека M2 и M5 подавляли активность RET, VEGFR1, VEGFR2, VEGFR3, KIT, PDGFR-alpha, PDGFR-beta, FGFR1, FGFR2, TIE2, DDR2, TrkA, Eph2A, RAF-1, BRAF, BRAF V600E, SAPK2, PTK5, Abl и CSF1R в концентрациях регорафениба, которые достигались клинически. В моделях in vivo регорафениб продемонстрировал антиангиогенную активность на модели опухоли у крыс и ингибирование роста опухоли на нескольких моделях ксенотрансплантата мышей, включая некоторые модели колоректальной карциномы человека, гастроинтестинальной стромальной и гепатоцеллюлярной карциномы. Регорафениб также продемонстрировал антиметастатическую активность на модели ксенотрансплантата мышей и двух ортотопических моделях колоректальной карциномы человека на мышах.
Фармакокинетика
Всасывание
После приема регорафениба в виде таблеток его средняя относительная биодоступность по сравнению с раствором для приема внутрь составляет 69–83%.
Среднее значение Сmax регорафениба в плазме крови составляет примерно 2,5 мг/л приблизительно через 3–4 ч после его однократного приема в дозе 160 мг.
Наибольшая концентрация регорафениба и его основных фармакологически активных метаболитов М2 (N-оксид) и М5 (N-оксид и N-дезметил) достигается при его приеме после завтрака с низким содержанием жиров в сравнении с применением после завтрака с высоким содержанием жиров или натощак. В сравнении с приемом натощак, экспозиция метаболитов М2 и М5 выше при приеме регорафениба после завтрака с низким содержанием жиров и ниже при приеме после завтрака с высоким содержанием жиров.
RxList.com (2016 г.)
Всасывание
После однократного приема регорафениба в дозе 160 мг больными с сóлидными опухолями среднее геометрическое значение Cmax регорафениба в плазме составляет 2,5 мкг/мл и достигается приблизительно в течение 4 ч, а среднее геометрическое значение AUC регорафениба — 70,4 мкг·ч/мл. Значение AUC регорафениба в равновесном состоянии возрастает менее чем прямо пропорционально при приеме более 60 мг. Средние геометрические значения Cmax и AUC регорафениба при равновесном состоянии достигают величин 3,9 мкг/мл и 58,3 мкг·ч/мл соответственно. Коэффициент вариации AUC и Cmax составляет от 35 до 44%.
Средняя относительная биодоступность регорафениба в виде таблеток по сравнению с раствором для перорального применения составляет 69–83%.
В исследовании по влиянию приема пищи на всасывание принимали участие 24 здоровых мужчины, принимавшие регорафениб однократно в дозе 160 мг натощак, с пищей с высоким содержанием жиров и низким содержанием жиров. Прием с пищей с высоким содержанием жиров (945 калорий и 54,6 г жиров) повышал среднее значение AUC регорафениба на 48% и снижал среднее значение AUC метаболитов М2 и М5 на 20 и 51% соответственно по сравнению с приемом натощак. Прием с пищей с низким содержанием жиров (319 ккал и 8,2 г жиров) повышал среднее значение AUC регорафениба, М2 и М5 на 36, 40 и 23% соответственно по сравнению с приемом натощак. В клинических исследованиях регорафениб принимали с пищей с низким содержанием жиров.
Распределение
Кривая зависимости концентрация-время демонстрирует несколько пиков как для регорафениба, так и для его основных циркулирующих метаболитов, в течение 24 ч после приема дозы, что связано с печеночно-кишечной рециркуляцией этого ЛС. Связь регорафениба с белками плазмы крови in vitro высокая и составляет 99,5%.
Метаболизм
Метаболизм регорафениба осуществляется главным образом в печени путем окисления, опосредованного изоферментом CYP3A4, а также путем глюкуронирования, опосредованного UGT1A9. В плазме выявляются 2 основных и 6 второстепенных метаболитов регорафениба. Основные циркулирующие в плазме крови метаболиты регорафениба М2 (N-оксид) и М5 (N-оксид и N-дезметил) обладают фармакологической активностью и в равновесном состоянии имеют концентрации, сходные с концентрацией регорафениба.
In vitro связь М2 и М5 с белками крови выше, чем у регорафениба, и составляет 99,8 и 99,95% соответственно.
Метаболиты могут быть восстановлены и гидролизированы микрофлорой ЖКТ, при этом возможно повторное всасывание неконъюгированного регорафениба и его метаболитов (печеночно-кишечная рециркуляция).
Выведение
Т1/2 регорафениба и его метаболита М2 из плазмы составляет 20–30 ч после приема внутрь. Средний Т1/2 метаболита М5 составляет »60 ч (40–100 ч).
Приблизительно 90% дозы радиоактивного регорафениба выводятся в течение 12 дней после его приема, при этом 71% выводится через кишечник (47% в виде исходного соединения и 24% в виде метаболитов) и около 19% — почками в виде глюкуронидов. В равновесном состоянии выведение глюкуронидов почками уменьшается и составляет меньше 10%. Исходное соединение, обнаруженное в каловых массах, может быть продуктом желудочно-кишечного расщепления глюкуронидов или продуктом восстановления метаболита М2 (N-оксида), или остатком неабсорбированного регорафениба.
RxList.com (2016 г.)
Выведение
После однократного приема регорафениба в дозе 160 мг среднее геометрическое значение (диапазон) Т1/2 регорафениба и метаболита М2 составляли 28 ч (14–58 ч) и 25 ч (14–32 ч) соответственно; у М5 было большее среднее значение (диапазон) Т1/2 — 51 ч (32–70 ч).
Около 71% радиоактивной метки выводилось с фекалиями (47% — исходное соединение, 24% — метаболиты) и 19% — мочой (17% — глюкурониды) в течение 12 дней после приема меченого регорафениба в виде раствора для приема внутрь в дозе 120 мг.
Линейность/нелинейность
Системное воздействие регорафениба при достижении Css возрастает пропорционально дозе при дозе <60 мг и менее пропорционально при дозах >60 мг. Накопление регорафениба при Css приблизительно в 2 раза превышает концентрацию в плазме, которая соответствует Т1/2 и частоте приема ЛС.
После приема регорафениба в дозе 160 мг его средняя Cmax в плазме крови в равновесном состоянии достигает 3,9 мг/л (8,1 мкмоль). Соотношение Cmax и Cmin регорафениба в плазме крови составляет менее 2.
Для обоих метаболитов М2 и М5 свойственно нелинейное накопление в плазме крови. После однократного приема регорафениба концентрации метаболитов М2 и М5 намного ниже, чем у исходного соединения. В равновесном состоянии концентрации М2 и М5 сопоставимы с концентрацией регорафениба.
Различные группы пациентов
Нарушение функции печени. У пациентов с легкой (класс А по классификации Чайлд-Пью) или умеренной (класс В по классификации Чайлд-Пью) степенью печеночной недостаточности фармакокинетические параметры регорафениба были такими же, как у пациентов с нормальной функцией печени. У пациентов с тяжелой степенью печеночной недостаточности (класс С по классификации Чайлд-Пью) фармакокинетика регорафениба не изучена. Поскольку печень имеет большое значение в выведении регорафениба, у пациентов с тяжелым нарушением функции печени возможно усиление действия этого ЛС (см. «Меры предосторожности»).
RxList.com (2016 г.)
Печеночная недостаточность. Фармакокинетические параметры регорафениба, его метаболитов М2 и М5 оценивались у 14 больных с гепатоцеллюлярной карциномой (ГЦК) и легкой печеночной недостаточностью (класс А по классификации Чайлд-Пью), у 4 больных с ГЦК и умеренной печеночной недостаточностью (класс В по классификации Чайлд-Пью) и у 10 пациентов с сóлидными опухолями и нормальной функцией печени после однократного приема регорафениба в дозе 100 мг. Клинически значимых различий значений средней экспозиции регорафениба, метаболитов М2 или М5 у пациентов с легкой или умеренной печеночной недостаточностью по сравнению с пациентами с нормальной функцией печени не наблюдалось. Фармакокинетика регорафениба у больных с тяжелой печеночной недостаточностью (класс С по классификации Чайлд-Пью) не изучена.
Нарушение функции почек. У пациентов с легкой и средней степенью почечной недостаточности экспозиция регорафениба и его метаболитов М2 и М5 в равновесном состоянии является такой же, как у пациентов с нормальной функцией почек. У пациентов с тяжелой степенью почечной недостаточности или терминальной степенью почечной недостаточности фармакокинетика регорафениба не изучена.
RxList.com (2016 г.)
Почечная недостаточность. Фармакокинетика регорафениба, его метаболитов М2 и М5 оценивалась у 10 больных с легкой степенью почечной недостаточности (Cl креатинина 60–89 мл/мин) и 18 пациентов с нормальной функцией почек после приема регорафениба в дозе 160 мг/сут в течение 21 дня. Каких-либо различий в значениях средней равновесной экспозиции регорафениба, метаболитов М2 или М5 у пациентов с легкой степенью почечной недостаточности по сравнению с пациентами с нормальной функцией почек не наблюдалось. Данные о фармакокинетике регорафениба у пациентов с умеренным нарушением функции почек (Cl креатинина 30–59 мл/мин) ограничены. Фармакокинетика регорафениба у больных с тяжелой почечной недостаточностью или терминальной стадией почечной недостаточности не исследовалась.
Пожилой возраст. Влияние возраста на фармакокинетику регорафениба у пациентов от 29 до 85 лет не обнаружено.
Пол. Не выявлено различий в фармакокинетике регорафениба в зависимости от пола.
Этническая принадлежность. Не выявлено различий в фармакокинетических параметрах регорафениба в зависимости от принадлежности к этнической группе.
RxList.com (обновление 2021 г.)
Возраст, пол, раса и масса тела не имели клинически значимого влияния на фармакокинетику регорафениба.
Печеночная недостаточность. На основании популяционного фармакокинетического анализа, не было отмечено клинически значимых различий в средней общей экспозиции регорафениба, включая M2 и M5, среди пациентов с нормальной функцией печени (общий билирубин и АСT ≤ВГН, n=744), легкой степенью печеночной недостаточности (общий билирубин ≤ВГН и AСT >ВГН или общий билирубин >ВГН до ≤1,5×ВГН, n=437) и умеренной печеночной недостаточностью (общий билирубин от >1,5 до ≤3×ВГН и любой АСT, n=36). Объединенный анализ включал 391 пациента с ГЦК, из которых 116, 249 и 26 были классифицированы как имеющие нормальную функцию печени, легкую и умеренную степень печеночной недостаточности соответственно. Фармакокинетика регорафениба не оценивалась у пациентов с тяжелой степенью печеночной недостаточности (общий билирубин >3×ВГН).
Почечная недостаточность. Фармакокинетика регорафениба, M2 и M5 была оценена у 6 пациентов с тяжелой степенью почечной недостаточности (Cl креатинина 15–29 мл/мин) и 18 пациентов с нормальной функцией почек/легкой степенью печеночной недостаточности (Cl креатинина ≥60 мл/мин) после приема регорафениба в дозе 160 мг ежедневно в течение 21 дня. Каких-либо различий в значениях экспозиции в равновесном состоянии регорафениба, М2 или М5 не наблюдалось у пациентов с тяжелой почечной недостаточностью по сравнению с пациентами с нормальной функцией почек. Фармакокинетика регорафениба не изучалась у пациентов с терминальной стадией почечной недостаточности, находящихся на диализе.
RxList.com (2016 г.)
Электрофизиология сердца. В открытом исследовании без контрольной группы у 25 пациентов с сóлидными опухолями изучалось влияние применения многократных доз регорафениба (160 мг 1 раз в день в течение 21 дня) на длину интервала QTc. Значимого удлинения интервала QTc (>20 мс) обнаружено не было.
Электрофизиология сердца/удлинение QT. У пациентов с онкологическими заболеваниями не было выявлено удлинение интервала QT в равновесном состоянии при приеме регорафениба в дозе 160 мг.
Применение вещества Регорафениб
Метастатический колоректальный рак у пациентов, которым уже проводилась или не показана химиотерапия фторпиримидиновыми ЛС, терапия, направленная против сосудистого эндотелиального фактора роста (VEGF), и терапия, направленная против рецепторов эпидермального фактора роста (EGFR) при диком типе KRAS; неоперабельные или метастатические гастроинтестинальные стромальные опухоли у пациентов при прогрессировании на терапии иматинибом и сунитинибом или при непереносимости данного вида лечения.
RxList.com (обновление 2021 г.)
Колоректальный рак. Регорафениб показан для лечения пациентов с метастатическим колоректальным раком, которые ранее получали химиотерапию фторпиримидином, оксалиплатином и иринотеканом, терапию против VEGF анти-EGFR терапию при диком типе RAS.
Гастроинтестинальные стромальные опухоли. Регорафениб показан для лечения пациентов с местнораспространенной, неоперабельной или метастатической стромальной гастроинтестинальной опухолью, которые ранее получали иматиниб и сунитиниб.
ГЦК. Регорафениб показан для лечения пациентов с ГЦК, которые ранее получали сорафениб.
Противопоказания
Повышенная чувствительность к регорафенибу; детский возраст до 18 лет; беременность и период грудного вскармливания (см. «Применение при беременности и кормлении грудью»); тяжелая степень печеночной недостаточности (класс С по классификации Чайлд-Пью); тяжелая степень почечной недостаточности (опыт клинического применения отсутствует).
RxList.com (обновление 2021 г.)
Нет.
Ограничения к применению
Нарушение функции печени легкой и средней степени тяжести; наличие мутации KRAS в опухоли; наличие факторов риска кровотечения, а также при совместном применении с антикоагулянтами и другими ЛС, повышающими риск кровотечений; ИБС.
Применение при беременности и кормлении грудью
Данные о применении регорафениба у беременных женщин отсутствуют. Учитывая механизм действия регорафениба, возможно его негативное влияние на плод. Исследования на животных продемонстрировали репродуктивную токсичность регорафениба.
Не установлено, выделяются ли регорафениб и его метаболиты с женским молоком. Исследования на животных показали, что регорафениб и его метаболиты выделяются с грудным молоком. Поскольку нельзя исключить возможность негативного влияния регорафениба на рост и развитие детей раннего возраста, следует прекратить грудное вскармливание в период лечения этим ЛС.
Фертильность. Не проводилось специальных исследований применения регорафениба для оценки его влияния на фертильность человека.
В исследованиях на животных наблюдалось снижение мужской и женской фертильности.
Женщин репродуктивного возраста необходимо проинформировать об опасности регорафениба для плода. Во время лечения и в течение 8 нед после терапии регорафенибом женщины и мужчины репродуктивного возраста должны применять надежные методы контрацепции.
RxList.com (2016 г.)
Категория действия на плод по FDA — D.
Основываясь на механизме действия регорафениба, это ЛС может нанести вред плоду при его применении беременной женщиной. Адекватные и хорошо контролируемые исследования применения регорафениба беременными женщинами отсутствуют. Регорафениб обладал эмбриолетальным и тератогенным действием у крыс и кроликов при более низких экспозициях, чем у человека при применении в рекомендуемой дозе, и вызывал возникновение пороков развития сердечно-сосудистой, мочеполовой и скелетно-мышечной систем с повышенной частотой. Если это ЛС применяется во время беременности или пациентка беременеет во время приема регорафениба, ее необходимо ознакомить с потенциальной опасностью его приема для плода.
Неизвестно, выводятся ли регорафениб и/или его метаболиты в материнское молоко. У крыс регорафениб и его метаболиты экскретируются в молоко из организма. В связи с тем, что многие ЛС выводятся в материнское молоко и из-за возможности возникновения тяжелых побочных реакций, вызываемых регорафенибом, у вскармливаемых грудью младенцев, должно быть принято решение о прекращении грудного вскармливания или приема регорафениба с учетом значения этого ЛС для матери.
RxList.com (обновление 2021 г.)
Применение при беременности
Предполагаемый фоновый риск серьезных врожденных дефектов и выкидыша у женщин неизвестен. В общей популяции в США оцениваемый фоновый уровень риска серьезных врожденных пороков развития и выкидыша при клинически установленной беременности составляет 2–4 и 15–20% соответственно.
Период лактации
Резюме рисков. Нет данных о наличии регорафениба или его метаболитов в материнском молоке, о влиянии регорафениба на грудного ребенка или на продукцию молока. У крыс регорафениб и его метаболиты экскретируются в молоко. Из-за возможности развития серьезных побочных реакций у детей, находящихся на грудном вскармливании, следует прекратить грудное вскармливание во время лечения регорафенибом и в течение 2 нед после последней дозы.
Побочные действия вещества Регорафениб
Общий профиль безопасности регорафениба оценивается по данным клинических исследований с участием более 1200 пациентов, получавших терапию в плацебо-контролируемых клинических исследованиях фазы 3. Данная группа включала 500 пациентов с метастатическим колоректальным раком и 132 пациента с гастроинтестинальными стромальными опухолями (ГСО).
В клинических исследованиях наиболее частыми нежелательными реакциями (≥30% пациентов) были астения/усталость, ладонно-подошвенная эритродизестезия (ЛПЭ), диарея, снижение аппетита и потребления пищи, повышение АД, дисфония, инфекции.
Наиболее серьезными нежелательными реакциями при приеме регорафениба были поражение печени, кровотечение, прободение ЖКТ.
Перечисленные ниже нежелательные явления, отмеченные при применении регорафениба в ходе клинических исследований, распределены по частоте возникновения в соответствии со следующей градацией: очень часто (≥1/10); часто (≥1/100, <1/10); нечасто (≥1/1000, <1/100); редко (≥1/10000, <1/1000). Для классификации и описания конкретной реакции, ее синонимов и связанных с ней состояний используется наиболее подходящий термин из MedDRA.
В каждой частотной группе нежелательные явления представлены в порядке уменьшения их значимости.
Со стороны крови и лимфатической системы: очень часто — тромбоцитопения, анемия; часто — лейкопения.
Со стороны сердца и сосудов: очень часто — кровотечения1, повышение АД; нечасто — инфаркт миокарда, ишемия миокарда, гипертонический криз.
Со стороны дыхательной системы, органов грудной клетки и средостения: очень часто — дисфония.
Со стороны кожи и подкожных тканей: очень часто — ЛПЭ, кожная сыпь, алопеция; часто — сухость кожи, эксфолиативный дерматит; нечасто — поражение ногтей, мультиформная эритема; редко — синдром Стивенса-Джонсона, токсический эпидермальный некролиз.
Со стороны ЖКТ: очень часто — диарея, стоматит, рвота, тошнота; часто — нарушение вкуса, сухость слизистой оболочки полости рта, гастроэзофагеальный рефлюкс, гастроэнтерит; нечасто — прободение ЖКТ1, свищ ЖКТ.
Со стороны печени и желчевыводящих путей: очень часто — гипербилирубинемия; часто — повышение активности трансаминаз; нечасто — тяжелое нарушение функции печени1,2.
Со стороны нервной системы: очень часто — головная боль; часто — тремор; редко — синдром задней обратимой энцефалопатии.
Со стороны скелетно-мышечной системы и соединительной ткани: часто — мышечно-скелетная ригидность.
Со стороны почек и мочевыводящих путей: часто — протеинурия.
Со стороны эндокринной системы: часто — гипотиреоз.
Со стороны обмена веществ и питания: очень часто — снижение аппетита и потребления пищи; часто — гипокалиемия, гипофосфатемия, гипокальциемия, гипонатриемия, гипомагниемия, гиперурикемия.
Лабораторные и инструментальные данные: очень часто — снижение массы тела; часто — увеличение активности амилазы и липазы, отклонение от нормального значения MHO.
Доброкачественные, злокачественные и неуточненные новообразования (включая кисты и полипы): редко — кератоакантома/плоскоклеточный рак кожи.
Инфекционные и паразитарные заболевания: очень часто — инфекции.
Общие расстройства и нарушения в месте введения: очень часто — астения/общая слабость, боль различной локализации, повышение температуры тела, воспаление слизистых оболочек.
1 Сообщалось о летальном исходе в результате неблагоприятной реакции.
2 В соответствии с критериями Международной рабочей группы экспертов по лекарственному поражению печени.
Поражение печени
Тяжелое лекарственное поражение печени со смертельным исходом наблюдалось у трех пациентов из более чем 1200, принимавших участие во всех клинических исследованиях и получавших терапию регорафенибом (0,25%). У двоих отмечались метастазы в печени. Дисфункция печени у данных пациентов началась в течение первых двух месяцев терапии и характеризовалась повреждением гепатоцитов с повышением активности трансаминаз 20×ВГН, сопровождаемым увеличением концентрации билирубина. В результате биопсии печени у двух пациентов был обнаружен некроз клеток печени с воспалительным инфильтратом.
Кровотечение
В двух плацебо-контролируемых исследованиях фазы 3 общая частота кровотечений у пациентов, получавших регорафениб, составила 19,3%. Большинство случаев кровотечения имели легкую или умеренную степень тяжести (1-я и 2-я степень: 16,9%). Наиболее часто отмечалось носовое кровотечение (7,6%). Летальные исходы у пациентов, принимавших регорафениб, отмечались редко (0,6%) и чаще были связаны с поражением дыхательной, пищеварительной и мочеполовой систем.
Инфекции
В двух плацебо-контролируемых исследованиях фазы 3 инфекционные заболевания чаще отмечались у пациентов, получавших лечение регорафенибом, по сравнению с больными, получавшими плацебо (все степени: 31% по сравнению с 14,4%). Чаще инфекции у пациентов, получавших регорафениб, имели легкую или умеренную степень выраженности (1-я и 2-я степень: 22,9%) и включали инфекции мочевыводящих путей (6,8%), назофарингит (4,2%), а также кандидоз кожи и слизистых и системный микоз (2,4%). Не наблюдалось различий по частоте летальных исходов в связи с развитием инфекции у пациентов, получавших регорафениб (0,6%), и у больных, получавших плацебо (0,6%).
В плацебо-контролируемом исследовании фазы 3 общая частота возникновения ЛПЭ у пациентов с метастатическим колоректальным раком, принимавших регорафениб, составила 45,2% и у пациентов, получающих плацебо, — 7,1%. В плацебо-контролируемом исследовании фазы 3 у пациентов с ГСО общая частота возникновения ЛПЭ составила 66,7% у пациентов, получавших терапию регорафенибом, и 15,2% у больных, получавших плацебо. В обоих исследованиях большинство случаев ЛПЭ у пациентов, получавших регорафениб, отмечалось во время первого цикла лечения и имело легкую или среднюю степень тяжести (1-я и 2-я степени: 28,6% у больных с метастатическим колоректальным раком и 44,7% у пациентов с ГСО). Частота возникновения ЛПЭ 3-й степени составила 16,6% у больных с метастатическим колоректальным раком и 22% у пациентов с ГСО.
Повышение АД
В плацебо-контролируемом исследовании фазы 3 у пациентов с метастатическим колоректальным раком общая частота случаев повышения АД составила 30,4% у пациентов, принимавших регорафениб, и 7,9% у пациентов, принимавших плацебо. В плацебо-контролируемом исследовании фазы 3 у пациентов с ГСО общая частота случаев повышения АД составила 59,1% у пациентов, получавших регорафениб, и 27,3% у пациентов, получавших плацебо. В обоих исследованиях большинство случаев повышения АД у пациентов, получавших регорафениб, было зарегистрировано в течение первого цикла лечения. При этом случаи повышения АД имели легкую и умеренную степень тяжести (1-я и 2-я степень: 22,8% у пациентов с метастатическим колоректальным раком и 31,1% у больных с ГСО). Частота случаев повышения АД 2-й степени составила 7,6% (у пациентов с колоректальным раком) и 27,3% (у пациентов с ГСО). Был зарегистрирован один случай повышения АД 4-й степени у пациента с ГСО.
Протеинурия
В плацебо-контролируемом исследовании фазы 3 среди пациентов с метастатическим колоректальным раком частота случаев протеинурии, связанной с лечением, составила 7,4% у пациентов, получавших терапию регорафенибом, в сравнении с 2,4% у пациентов, принимавших плацебо. После развития протеинурии у 40,5% пациентов из группы регорафениба и 66,7% пациентов из группы плацебо состояние не вернулось к исходным значениям. В плацебо-контролируемом исследовании фазы 3 среди пациентов с ГСО общая частота случаев протеинурии составила 6,8% у пациентов, получающих терапию регорафенибом, в сравнении с 1,5% у больных, принимавших плацебо.
Нарушения со стороны сердца и сосудов
Во всех проводимых клинических исследованиях нежелательные явления в виде нарушений со стороны сердца (все степени) чаще регистрировались (20,5% относительно 10,4 %) у пациентов, получавших терапию регорафенибом, в возрасте 75 лет или старше (n=78), чем у пациентов, получавших терапию регорафенибом, в возрасте до 75 лет (n=995).
Лабораторные и инструментальные данные
В двух плацебо-контролируемых исследованиях фазы 3 у 26,1% пациентов, получавших терапию регорафенибом, и у 15,1% больных, получавших плацебо, концентрация ТТГ была выше ВГН. Значения ТТГ, в 4 раза превышающие ВГН, были зарегистрированы у 6,9% пациентов, получавших терапию регорафенибом, и у 0,7% пациентов, принимавших плацебо. Концентрация свободного трийодтиронина (Т3 свободный) меньше НГН отмечалась у 25,6% пациентов, получающих терапию регорафенибом, и у 20,9% пациентов, получающих плацебо. Концентрация свободного тироксина (Т4 свободный) меньше НГН отмечалась у 8% пациентов в группе терапии регорафенибом и у 6,6% пациентов из группы плацебо. В целом приблизительно у 7% пациентов, получающих терапию регорафенибом, развился гипотиреоз, требующий применения заместительной гормонотерапии.
RxList.com
Так как клинические испытания проведены с различным набором условий, частота встречаемости побочных реакций, наблюдавшихся в этих исследованиях, может не совпадать с полученной в других исследованиях и с наблюдаемой в клинической практике.
Наиболее часто наблюдаемыми побочными реакциями (≥20%) у пациентов, принимавшими регорафениб, являются астения/усталость; ЛПЭ; диарея; снижение аппетита/потребления пищи; гипертония; мукозит; дисфония; инфекции; боль; снижение веса; боли в животе; сыпь; лихорадка и тошнота.
Наиболее тяжелыми побочными реакциями у пациентов, принимающих регорафениб, являются гепатотоксичность; кровотечение; прободение ЖКТ.
Опыт клинических испытаний
Колоректальный рак. Описанные ниже данные по исследованию безопасности, за исключением особо оговоренных случаев, получены в рандомизированном (2:1) двойном слепом плацебо-контролируемом исследовании (исследование 1), в котором 500 пациентов, ранее проходивших лечение (средний возраст 61 год, 61% — мужчины), с метастатическим колоректальным раком, принимали регорафениб в качестве единственного ЛС в дозе 160 мг/сут в течение первых 3 нед каждого 4-недельного цикла лечения, и 253 пациента (средний возраст 61 год, 60% — мужчины) принимали плацебо. Средняя продолжительность терапии составила 7,3 нед (диапазон 0,3; 47) у пациентов, принимавших регорафениб. В связи с развитием побочных реакций у 61% пациентов, принимавших регорафениб, потребовалось прерывание лечения и у 38% пациентов — снижение дозирования. Лекарственноиндуцированные побочные реакции, развитие которых привело к прекращению лечения, были зарегистрированы у 8,2% больных, принимавших регорафениб, по сравнению с 1,2% пациентов, получавших плацебо. ЛПЭ и сыпь были наиболее частыми причинами прекращения приема регорафениба.
Сравнение частоты возникновения побочных реакций (≥10%) у пациентов, принимавших регорафениб (n=500, все степени тяжести в %, ≥3-й степени тяжести в % — в скобках) и возникавших чаще, чем у пациентов, получавших плацебо (n=253, все степени тяжести в %, ≥3-й степени тяжести в % — в скобках) в исследовании 1 представлено ниже.
Общие расстройства и нарушения в месте введения: астения/усталость — 64% (15%) и 46% (9%); боль — 29% (3%) и 21% (2%); лихорадка — 28% (2%) и 15% (0%).
Со стороны обмена веществ и питания: снижение аппетита и потребления пищи — 47% (5%) и 28% (4%).
Со стороны кожи и подкожной клетчатки: ЛПЭ — 45% (17%) и 7% (0%); сыпь1 — 26% (6%) и 4% (<1%).
Со стороны ЖКТ: диарея — 43% (8%) и 17% (2%); мукозит — 33% (4%) и 5% (0%).
Исследования: потеря веса — 32% (<1%) и 10% (0%).
Инфекции и инвазии: инфекция — 31% (9%) и 17% (6%).
Со стороны ССС: гипертония — 30% (8%) и 8% (<1%); кровотечение2 — 21% (2%) и 8% (<1%).
Со стороны дыхательной системы, органов грудной клетки и средостения: дисфония — 30% (0%) и 6% (0%).
Со стороны нервной системы: головная боль — 10% (<1%) и 7% (0%).
Отклонения лабораторных показателей. Сравнение частоты возникновения отклонений лабораторных показателей у больных, принимавших регорафениб (n=5003, все степени тяжести4 в %, 3-й и 4-й степени тяжести4 в % — в скобках), и пациентов, получавших плацебо (n=2533, все степени тяжести4 в %, 3-й и 4-й степени тяжести4 в % — в скобках), в исследовании 1 представлено ниже.
Со стороны крови и лимфатической системы: анемия — 79% (5 и 1%) и 66% (3 и 0%); тромбоцитопения — 41% (2 и <1%) и 17% (<1 и 0%); нейтропения — 3% (1 и 0%) и 0% (0 и 0%); лимфопения — 54% (9 и 0%) и 34% (3 и 0%).
Со стороны обмена веществ и питания: гипокальциемия — 59% (1 и <1%) и 18% (1 и 0%); гипокалиемия — 26% (4 и 0%) и 8% (<1 и 0%); гипонатриемия — 30% (7 и 1%) и 22% (4 и 0%); гипофосфатемия — 57% (31 и 1%) и 11% (4 и 0%).
Со стороны печени и желчевыводящих путей: гипербилирубинемия — 45% (10 и 3%) и 17% (5 и 3%); повышение активности АСТ — 65% (5 и 1%) и 46% (4 и 1%); повышение активности AЛT — 45% (5 и 1%) и 30% и (3 и <1%).
Со стороны почек и мочевыводящих путей: протеинурия — 60% (<1 и 0%) и 34% (<1 и 0%).
Исследования: повышение значения МНО5 — 24% (4% и Н/Д) и 17% (2% и Н/Д); повышение активности липазы — 46% (9 и 2%) и 19% (3 и 2%); повышение активности амилазы — 26% (2 и <1%) и 17% (2 и <1%).
1 Термин сыпь включает в себя наблюдавшиеся случаи лекарственной сыпи, в т.ч. эритематозной, генерализованной, макулярной, пятнисто-папулезной, папулезной, зудящей.
2 Наблюдались летальные исходы.
3 % — в зависимости от количества пациентов с исследованными лабораторными показателями, которых могло быть меньше, чем 500 (регорафениб) или 253 (плацебо).
4 На основании Общих терминологических критериев нежелательных явлений — Commom Terminology Criteria for Adverse Events (CTCAE), версия 3.0.
5 МНО: значение 4-й степени тяжести на основании CTCAE версии 3.0 не определяется.
ГСО. Описанные ниже данные по безопасности получены в рандомизированном (2:1) двойном слепом плацебо-контролируемом исследовании (исследование 2), в котором принимали участие 132 пациента (средний возраст 60 лет, 64% — мужчины) с предварительным лечением ГСО, принимавших только регорафениб в дозе 160 мг в день в течение первых 3 нед каждого 4-недельного цикла лечения, и 66 пациентов (средний возраст 61 год, 64% — мужчины), принимавших плацебо. Средняя продолжительность терапии составила 22,9 нед (диапазон 0,1; 50,9) для пациентов, принимавших регорафениб. В связи с развитием побочных реакций у 58% пациентов, принимавших регорафениб, потребовалось прерывание лечения и у 50% пациентов — снижение дозирования. Лекарственноиндуцированные побочные реакции, развитие которых привело к прекращению лечения, были зарегистрированы у 2,3% больных, принимавших регорафениб, по сравнению с 1,5% пациентов, получавших плацебо.
Сравнение частоты развития побочных реакций, возникавших чаще (≥10%) у пациентов, принимавших регорафениб (n=132, все степени тяжести в %, ≥3-й степени тяжести в % — в скобках), чем у пациентов, получавших плацебо (n=66, все степени тяжести в %, ≥3-й степени тяжести в % — в скобках) в исследовании 2 представлено ниже.
Со стороны кожи и подкожной клетчатки: ЛПЭ — 67% (22%) и 12% (2%); сыпь6 — 30% (7%) и 3% (0%); алопеция — 24% (2%) и 2% (0%).
Общие расстройства и нарушения в месте введения: астения/усталость — 52% (4%) и 39% (2%); лихорадка — 21% (0%) и 11% (2%).
Со стороны ССС: гипертония — 59% (28%) и 27% (5%); кровотечение — 11% (4%) и 3% (0%).
Со стороны ЖКТ: диарея — 47% (8%) и 9% (0%); мукозит — 40% (2%) и 8% (2%); тошнота — 20% (2%) и 12% (2%); рвота — 17% (<1%) и 8% (0%).
Со стороны дыхательной системы, органов грудной клетки и средостения: дисфония — 39% (0%) и 9% (0%).
Инфекции и инвазии: инфекция — 32% (5%) и 5% (0%).
Со стороны обмена веществ и питания: снижение аппетита и потребления пищи — 31% (<1%) и 21% (3%); гипотиреоз7 — 18% (0%) и 6% (0%).
Со стороны нервной системы: головная боль — 16% (0%) и 9% (0%).
Исследования: потеря веса — 14% (0%) и 8% (0%).
Со стороны скелетно-мышечной и соединительной ткани: скелетно-мышечная ригидность — 14% (0%) и 3% (0%).
6 Термин сыпь включает в себя наблюдавшиеся случаи, в т.ч. эритематозной, макулярной, пятнисто-папулезной, папулезной и зудящей сыпи.
7 Случаи гипотериоза, выявленные в подгруппе пациентов с исходными (до лечения) нормальными уровнем ТТГ и функцией щитовидной железы.
Отклонения лабораторных показателей. Сравнение частоты возникновения отклонений лабораторных показателей у больных, принимавших регорафениб (n=1328, все степени тяжести9 в %, 3-й и 4-й степени тяжести9 в % — в скобках), и пациентов, получавших плацебо (n=668, все степени тяжести9 в %, 3-й и 4-й степени тяжести9 в % — в скобках), в исследовании 2 представлено ниже.
Со стороны крови и лимфатической системы: тромбоцитопения — 13% (1 и 0%) и 2% (0 и 2%); нейтропения — 16% (2 и 0%) и 12% (3 и 0%); лимфопения — 30% (8 и 0%) и 24% (3 и 0%).
Со стороны обмена веществ и питания: гипокальцемия — 17% (2 и 0%) и 5% (0 и 0%); гипокалиемия — 21% (3 и 0%) и 3% (0 и 0%); гипофосфатемия — 55% (20 и 2%) и 3% (2 и 0%).
Со стороны печени и желчевыводящих путей: гипербилирубинемия — 33% (3 и 1%) и 12% (2 и 0%); повышение активности АСТ — 58% (3 и 1%) и 47% (3 и 0%); повышение активности АЛТ — 39% (4 и 1%) и 39% (2 и 0%).
Со стороны почек и мочевыводящих путей: протеинурия10 — 33% (3% и Н/Д) и 30% (3% и Н/Д).
Исследования: повышение активности липазы — 14% (0 и 1%) и 5% (0 и 0%).
8 % — в зависимости от количества пациентов с исследованными лабораторными показателями, которых могло быть меньше чем 132 (регорафениб) или 66 (плацебо).
9 На основании CTCAE, версия 4.0.
10 значение 4-й степени тяжести на основании CTCAE версии 4.0 не определяется.
Опыт постмаркетинговых исследований
Побочное действие, выявленное в ходе пострегистрационного применения регорафениба, приводится далее. В связи с тем, что данные об этой реакции добровольно поступают от населения неопределенной численности, не всегда можно достоверно оценить частоту ее возникновения или установить причинно-следственную связь с экспозицией исследуемого ЛС.
Реакция гиперчувствительности.
RxList.com (обновление 2021 г.)
Следующие серьезные побочные реакции обсуждаются в другом разделе:
— гепатотоксичность (см. «Меры предосторожности»);
— инфекции (см. «Меры предосторожности»);
— кровотечение (см. «Меры предосторожности»);
— прободение или свищ ЖКТ (см. «Меры предосторожности»);
— кожная токсичность (см. «Меры предосторожности»);
— гипертензия (см. «Меры предосторожности»);
— ишемия и инфаркт миокарда (см. «Меры предосторожности»);
— синдром обратимой задней энцефалопатии (см. «Меры предосторожности»).
Опыт клинических испытаний
Поскольку клинические испытания проведены с различным набором условий, частота встречаемости побочных реакций, наблюдавшихся в этих исследованиях, может не совпадать с полученной в других исследованиях и с наблюдаемой в клинической практике.
Данные, описанные в разделе «Меры предосторожности», отражают экспозицию регорафениба у более чем 4800 пациентов, которые были включены в 4 рандомизированных плацебо-контролируемых исследования (n=1142), программу расширенного доступа (CONSIGN, n=2864) или несравнительное клиническое испытание (одно ЛС или в комбинации с другими ЛС). 4518 пациентов получали регорафениб в качестве единственного ЛС; распределение основных злокачественных новообразований было следующим: 80% — колоректальный рак, 4% — ГСО, 10% — ГЦК, 6% — другие сóлидные опухоли. 74% пациентов были белокожими, 11% — азиатами и у 15% — раса была неизвестна. Среди этих 4518 пациентов 83% получали регорафениб в течение как минимум 21 дня, а 20% — в течение 6 мес или дольше.
В рандомизированных плацебо контролируемых исследованиях (CORRECT, GRID, RESORCE и CONCUR) наиболее часто наблюдаемыми побочными реакциями (≥20%) у пациентов, принимавших регорафениб, являются боль (включая боль в ЖКТ и абдоминальную боль), ладонно-подошвенная эритродизестезия, астения/усталость, диарея, снижение аппетита/приема пищи, гипертензия, инфекции, дисфония, гипербилирубинемия, лихорадка, мукозит, потеря веса, сыпь и тошнота.
Колоректальный рак
Таблица 1
Побочные реакции, зарегистрированные у ≥10% пациентов, получавших регорафениб в исследовании CORRECT, и возникавшие чаще, чем у пациентов, получавших плацебо1
| Побочная реакция | Регорафениб (n=500) | Плацебо (n=253) | ||
| Степень | Степень | |||
| Все % | ≥3% | Все % | ≥3% | |
| Общие расстройства и нарушения в месте введения | ||||
| Боль | 59 | 9 | 48 | 7 |
1 Побочные реакции классифицированы в соответствии с Общими критериями токсичности Национального института рака США — National Cancer Institute Common Toxicity for Adverse Events (NCI CTCAE), версия 3.0.
Таблица 2
Отклонения некоторых лабораторных показателей, зафиксированные в исследовании CORRECT
| Лабораторный параметр | Регорафениб (n=5001) | Плацебо (n=2531) | ||||
| Степень2 | Степень2 | |||||
| Все % | 3% | 4% | Все % | 3% | 4% | |
| Нарушения со стороны крови и лимфатической системы | ||||||
| Лимфопения | 54 | 9 | 0 | 35 | 4 | <1 |
| Со стороны почек и мочевыводящих путей | ||||||
| Протеинурия3 | 84 | 2 | 0 | 61 | 1 | 0 |
1 % в зависимости от количества пациентов с исследованными лабораторными показателями после исходного уровня, которых могло быть меньше, чем 500 (регорафениб) или 253 (плацебо).
2 NCI CTCAE, версия 3.0.
3 На основе данных о соотношении белок/креатинин в моче.
ГСО
Таблица 3
Побочные реакции, зарегистрированные у ≥10% пациентов, получавших регорафениб в исследовании GRID, и возникавшие чаще, чем у пациентов, получавших плацебо1
| Побочная реакция | Регорафениб (n=132) | Плацебо (n=66) | ||
| Степень | Степень | |||
| Все % | ≥3% | Все % | ≥3% | |
| Общие расстройства и нарушения в месте введения | ||||
| Боль | 60 | 8 | 55 | 14 |
1 Побочные реакции классифицированы в соответствии c NCI CTCAE, версия 4.0.
Таблица 4
Отклонения некоторых лабораторных показателей, зафиксированные в исследовании GRID
| Лабораторный параметр | Регорафениб (n=1321) | Плацебо (n=661) | ||||
| Степень2 | Степень2 | |||||
| Все % | 3% | 4% | Все % | 3% | 4% | |
| Нарушения со стороны крови и лимфатической системы | ||||||
| Нейтропения | 16 | 2 | 1 | 12 | 3 | 0 |
| Со стороны почек и мочевыводящих путей | ||||||
| Протеинурия3 | 59 | 3 | —4 | 53 | 3 | —4 |
1 % в зависимости от количества пациентов с исследованными лабораторными показателями после исходного уровня, которых могло быть меньше, чем 132 (регорафениб) или 66 (плацебо).
2 NCI CTCAE, версия 3.0.
3 На основе данных о соотношении белок/креатинин в моче.
4 На основе NCI CTCAE версия 4.0 оценка 4 отсутствует.
ГЦК
Описанные ниже данные по безопасности получены в рандомизированном (2:1) двойном слепом плацебо-контролируемом исследовании (RESORCE), в котором пациенты с ранее леченной ГЦК получали либо регорафениб (n=374) 160 мг перорально в 1–21-й день каждого 4-недельного цикла лечения, либо плацебо (n=193). Средний возраст составлял 63 года, 88% — мужчины, у 98% был цирроз печени (Чайлд-Пью A), 66% имели 0 баллов по шкале ECOG (шкала оценки общего состояния онкологического пациента — Eastern Cooperative Oncology Group, ECOG) и 34% — 1 балл. Средняя продолжительность терапии составляла 3,5 мес (от 1 дня до 29,4 мес) для пациентов, получающих регорафениб, из них 33% принимали регорафениб в течение ≥6 мес, а 14% — в течение ≥12 мес. Прерывание лечения из-за развития побочных эффектов потребовалось у 58,3% пациентов, получавших регорафениб, и у 48% пациентов доза была снижена. Наиболее частыми побочными реакциями, требующими изменения дозы (прерывание или снижение дозы), были ладонно-подошвенная кожная реакция (hand-foot skin reaction, HFSR, ЛПР)/ладонно-подошвенная (палмарно-плантарная) эритродизестезия (palmar-plantar erythrodysesthesia (PPES, ЛПЭ) (1,9%) и повышение уровня АСТ (1,6%).
В таблице 5 представлена частота возникновения побочных реакций (≥10%) у пациентов в исследовании RESORCE.
Таблица 5
Побочные реакции, зарегистрированные у ≥10% пациентов, получавших регорафениб в исследовании RESORCE, и возникавшие чаще, чем у пациентов, получавших плацебо1
| Побочная реакция | Регорафениб (n=374) | Плацебо (n=193) | ||
| Степень | Степень | |||
| Все % | ≥3% | Все % | ≥3% | |
| Заболевания кожи и подкожной клетчатки | ||||
| ЛПР/ЛПЭ | 51 | 12 | 7 | <1 |
| Общие расстройства и нарушения в месте введения | ||||
| Боль | 55 | 9 | 44 | 8 |
| Астения/усталость | 42 | 10 | 33 | 5 |
| Лихорадка | 20 | 0 | 7 | 0 |
| Сосудистые расстройства | ||||
| Гипертензия | 31 | 15 | 6 | 5 |
| Кровотечение2 | 18 | 5 | 16 | 8 |
| Желудочно-кишечные расстройства | ||||
| Диарея | 41 | 3 | 15 | 0 |
| Тошнота | 17 | <1 | 13 | 0 |
| Рвота | 13 | <1 | 7 | <1 |
| Мукозит | 13 | 1 | 2 | <1 |
| Расстройства со стороны дыхательной системы, органов грудной клетки и средостения | ||||
| Дисфония | 18 | 0 | 2 | 0 |
| Инфекции и инвазии | ||||
| Инфекция2 | 31 | 8 | 18 | 6 |
| Нарушения обмена веществ и питания | ||||
| Снижение аппетита и потребления пищи | 31 | 3 | 15 | 2 |
| Исследования | ||||
| Потеря массы тела | 13 | 2 | 4 | 0 |
| Нарушения со стороны скелетно-мышечной системы и соединительной ткани | ||||
| Мышечные спазмы | 10 | 0 | 2 | 0 |
1 Побочные реакции классифицированы в соответствии с NCI CTCAE, версия 4.0.
2 Наблюдались летальные исходы.
Другими клинически значимыми побочными реакциями, наблюдавшимися у <10% пациентов, получавших регорафениб, были алопеция (7%), гипотиреоз (6,4%), панкреатит (1,6%), эксфолиативная сыпь (1,3%), тремор (1,3%), многоформная эритема (0,8%), ишемия миокарда (0,8%), желудочно-кишечный свищ (0,3%) и инфаркт миокарда (0,3%).
В таблице 6 представлены лабораторные отклонения, наблюдавшиеся в исследовании RESORCE.
Таблица 6
Отклонения некоторых лабораторных показателей, зафиксированные в исследовании RESORCE
| Лабораторный параметр | Регорафениб (n=3741) | Плацебо (n=1931) | ||||
| Степень2 | Степень2 | |||||
| Все % | 3% | 4% | Все % | 3% | 4% | |
| Нарушения со стороны крови и лимфатической системы | ||||||
| Тромбоцитопения | 63 | 5 | <1 | 50 | 0 | 0 |
| Нейтропения | 14 | 3 | 0 | 15 | <1 | <1 |
| Лимфопения | 68 | 16 | 2 | 59 | 11 | <1 |
| Нарушения обмена веществ и питания | ||||||
| Гипокальциемия | 23 | <1 | 0 | 10 | 0 | 0 |
| Гипокалиемия | 31 | 4 | <1 | 9 | 2 | 0 |
| Гипофосфатемия | 70 | 32 | 2 | 31 | 7 | 0 |
| Гепатобилиарные нарушения | ||||||
| Гипербилирубинемия | 78 | 13 | 3 | 55 | 11 | 5 |
| Повышение АСТ | 93 | 16 | 2 | 84 | 17 | 3 |
| Повышение АЛТ | 70 | 6 | <1 | 59 | 5 | 0 |
| Со стороны почек и мочевыводящих путей | ||||||
| Протеинурия3 | 51 | 17 | —3 | 37 | 3 | —3 |
| Исследования | ||||||
| Повышение МНО | 44 | <1 | —4 | 35 | 2 | —4 |
| Повышение липазы | 41 | 11 | 3 | 27 | 8 | 1 |
| Повышение амилазы | 23 | 3 | <1 | 19 | 2 | <1 |
1 % в зависимости от количества пациентов с исследованными лабораторными показателями после исходного уровня, которых могло быть меньше, чем 374 (регорафениб) или 193 (плацебо).
2 NCI CTCAE, версия 4.0.
3 На основе определения.
4 На основе NCI CTCAE версия 4.0 оценка 4 отсутствует.
Опыт постмаркетинговых исследований
В ходе пострегистрационного применения регорафениба выявлены следующие побочные эффекты:
— реакция гиперчувствительности;
— нефротический синдром;
— сердечная недостаточность;
— артериальные (включая аортальные) аневризмы, расслоения и разрывы.
Взаимодействие
RxList.com
Влияние регорафениба на субстраты цитохрома Р450. Исследования in vitro показали, что регорафениб является ингибитором изоферментов CYP2C8, CYP2C9, CYP2B6, CYP3A4 и CYP2C19; метаболит М2 регорафениба является ингибитором изоферментов CYP2C9, CYP2C8, CYP3A4 и CYP2D6 и метаболит М5 — ингибитор изофермента CYP2C8. По данным исследований in vitro, регорафениб не является индуктором изоферментов CYP1A2, CYP2B6, CYP2C19 и CYP3A4.
Фармакокинетические взаимодействия
Индукторы изофермента СYР3А4/ингибиторы изофермента CYP3A4 и изофермента UGT1A9
In vitro было показано, что регорафениб метаболизируется изоферментом CYP3A4 и изоферментом UGT1А9.
Применение кетоконазола (400 мг в течение 18 дней), сильного ингибитора изофермента CYP3A4, в сочетании с однократным приемом регорафениба (160 мг на 5-й день) приводило к увеличению значения AUC регорафениба на »33% и снижению среднего значения AUC его активных метаболитов, М2 (N-оксид) и М5 (N-оксид и N-дезметил), на »90%. Не рекомендуется применять регорафениб совместно с сильными ингибиторами изофермента CYP3A4 (например кларитромицин, грейпфрутовый сок, итраконазол, кетоконазол, позаконазол, телитромицин и вориконазол), поскольку их влияние на воздействие регорафениба в стабильном состоянии и его метаболитов (М2 и М5) не изучалось.
RxList.com (обновление 2021 г.)
Влияние сильных ингибиторов CYP3A4 на регорафениб
Исследование взаимодействия кетоконазола и регорафениба было проведено с участием 18 здоровых мужчин. Средняя AUC регорафениба увеличилась на 33%, а средняя AUC M2 и M5 снизилась на 93%.
Не рекомендуется применять сильные ингибиторы изофермента UGT1A9 (например мефенаминовая кислота, дифлунизал и нифлумовая кислота) во время терапии регорафенибом, поскольку их влияние на экспозицию регорафениба и его метаболитов в равновесном состоянии не изучалось.
Применение рифампицина (600 мг в течение 9 дней), сильного индуктора изофермента CYP3A4, в сочетании с однократным приемом регорафениба (160 мг на 7-й день) приводило к снижению значения AUC регорафениба приблизительно на 50%, увеличению среднего значения AUC активного метаболита М5 в 3–4 раза, при этом не отмечалось изменения экспозиции активного метаболита М2. Другие сильные индукторы изофермента CYP3A4 (например фенитоин, карбамазепин, фенобарбитал) могут увеличивать метаболизм регорафениба. Не рекомендуется применять регорафениб совместно с сильными индукторами изофермента CYP3A4 или подбирать ЛС, которые не влияют на CYP3A4 или индуцируют его в минимальной степени.
RxList.com (обновление 2021 г.)
Влияние сильных индукторов CYP3A4 на регорафениб
Исследование взаимодействия рифампицина и регорафениба было проведено с участием 22 здоровых мужчин. Средняя AUC регорафениба снизилась на 50%, а средняя AUC M5 увеличилась на 264%. Изменений средней AUC M2 не наблюдалось.
Совместное применение сильного индуктора CYP3A4 с регорафенибом снижает плазменные концентрации регорафениба, увеличивает плазменные концентрации активного метаболита M5 и не приводит к изменению плазменных концентраций активного метаболита M2, и может привести к снижению эффективности. Следует избегать одновременного применения регорафениба с сильными индукторами CYP3A4 (например, рифампицин, фенитоин, карбамазепин, фенобарбитал и зверобой).
Субстраты изоферментов UGT1A1 и UGT1A9
In vitro было показано, что регорафениб, также как и его активный метаболит М2, подавляет глюкуронизацию под действием изоферментов UGT1А1 и UGT1А9, в то время как метаболит М5 подавляет изофермент UGT1A1 только в концентрациях, которые достигаются в равновесном состоянии in vivo.
Применение регорафениба с последующим 5-дневным перерывом перед назначением иринотекана приводило к увеличению среднего значения AUC SN-38 , субстрата изофермента UGT1А1 и активного метаболита иринотекана приблизительно на 44%. Также отмечалось увеличение среднего значения AUC иринотекана приблизительно на 28%. Эти данные показывают, что комбинированное применение регорафениба может увеличивать системную экспозицию субстратов изоферментов UGT1А1 и UGT1A9.
RxList.com (обновление 2021 г.)
Влияние регорафениба на субстраты UGT1A1
Исследования in vitro показали, что регорафениб, М2 и М5 конкурентно ингибируют UGT1A9 и UGT1A1 в терапевтически релевантных концентрациях. Одиннадцать пациентов прошли комбинированную химиотерапию с иринотеканом при применении регорафениба в дозе 160 мг. Средняя AUC иринотекана увеличилась на 28%, а средняя AUC SN-38 увеличилась на 44%, когда иринотекан применяли через 5 дней после последней из 7-дневных доз регорафениба.
Субстраты BCRP и Р-gp
In vitro было показано, что регорафениб (IC50 около 40–70 нмоль) и его метаболиты М2 (IC50=390 нмоль) и М5 (IC50=150 нмоль) являются ингибиторами BCRP. Регорафениб (IC50 около 2 мкмоль) и его метаболит М2 (IC50=1,5 мкмоль) ингибируют Р-gp. Комбинированное применение с регорафенибом может повышать концентрацию в плазме сопутствующих субстратов BCRP (например метотрексат) или субстратов Р-gp (например дигоксин).
RxList.com (обновление 2021 г.)
Влияние регорафениба на субстраты BCRP
Совместное применение регорафениба с субстратом BCRP увеличивало плазменные концентрации субстрата BCRP. Необходим тщательный мониторинг пациентов на предмет обнаружения признаков и симптомов токсичности, связанной с воздействием на субстрат BCRP (например, метотрексат, флувастатин, аторвастатин). Необходимо ознакомиться с сопутствующей информацией о субстрате BCRP при рассмотрении вопроса о применении таких ЛС вместе с регорафенибом.
Прием регорафениба (160 мг в течение 14 дней) перед применением разовой дозы розувастатина (5 мг), субстрата BCRP, привело к увеличению средней экспозиции (AUC) розувастатина в 3,8 раза и увеличению его Cmax в 4,6 раза.
Ингибиторы Р-gp и BCRP/индукторы Р-gp и BCRP
Исследования in vitro показывают, что метаболиты регорафениба М2 и М5 являются субстратами Р-gp и BCRP. Ингибиторы и индукторы BCRP и Р-gp могут изменять экспозицию М2 и М5. Клиническая значимость данных результатов исследований неизвестна.
Селективные субстраты изоферментов CYP
In vitro было показано, что регорафениб является конкурентоспособным ингибитором изоферментов CYP2C8, CYP2C9, CYP2B6 в концентрациях, которые достигаются in vivo в стабильном состоянии (значение Cmax в плазме составляет 8,1 мкмоля). In vitro ингибирующее действие в отношении изоферментов CYP3A4 и CYP2C19 менее выражено. Было проведено исследование с целью оценки влияния применения регорафениба в дозе 160 мг в течение 14 дней на фармакокинетику маркерных субстратов изоферментов CYP2C8 (росиглитазон), CYP2C9 (варфарин), CYP2C19 (омепразол) и CYP3A4 (мидазолам). Фармакокинетические данные показывают, что регорафениб можно применять совместно с субстратами изоферментов CYP2C8, CYP2C9, CYP3A4 и CYP2C19. При этом не отмечается клинически значимое взаимодействие между ЛС.
RxList.com (обновление 2021 г.)
Влияние регорафениба на субстраты цитохрома P450
Пациенты с сóлидными опухолями получали однократные пероральные дозы субстратов CYP: 2 мг мидазолама (CYP3A4), 40 мг омепразола (CYP2C19) и 10 мг варфарина (CYP2C9) или 4 мг росиглитазона (CYP2C8) за 1 нед до и через 2 нед после регорафениба в дозе 160 мг 1 раз в сутки. Не наблюдалось клинически значимого эффекта в отношении средней AUC розиглитазона (n=12) или средней концентрации омепразола в плазме (n=11), измеренных через 6 ч после приема, или средней AUC мидазолама (n=15). Средняя AUC варфарина (n=8) увеличилась на 25%.
Антибиотики
Профиль концентрация-время показывает, что регорафениб и его метаболиты могут подвергаться печеночно-кишечной циркуляции (см. «Фармакология»). Комбинированное применение антибиотиков, влияющих на флору ЖКТ, может нарушить печеночно-кишечную циркуляцию регорафениба и привести к уменьшению его экспозиции. Клиническая значимость данного взаимодействия неизвестна, но может являться причиной снижения эффективности регорафениба.
RxList.com (обновление 2021 г.)
Влияние неомицина на регорафениб
Двадцать семь здоровых мужчин получили однократную дозу регорафениба 160 мг через 5 дней после начала приема неомицина. Неомицин, неабсорбируемый антибиотик, применяли в дозе 1 г трижды в день в течение 5 дней. Клинически значимого влияния на среднюю AUC регорафениба не наблюдалось; однако средняя AUC M2 снизилась на 76%, а средняя AUC M5 уменьшилась на 86%. Уменьшение экспозиции M2 и M5 может привести к снижению эффективности регорафениба. Влияние других антибиотиков на воздействие регорафениба и его активных метаболитов не изучалось.
Комплексообразующие соединения солей желчных кислот
Существует вероятность, что регорафениб и его метаболиты М2 и М5 могут подвергаться печеночно-кишечной циркуляции (см. «Фармакология»). Комплексообразующие соединения солей желчных кислот, такие как холестирамин и холестагель (колесевелам), могут взаимодействовать с регорафенибом, образуя нерастворимые комплексы, которые оказывают влияние на абсорбцию (или реабсорбцию), что потенциально может привести к снижению экспозиции. Клиническое значение данных потенциальных взаимодействий неизвестно, но может привести к снижению эффективности регорафениба.
Передозировка
Симптомы: в клинических исследованиях максимальная суточная доза регорафениба составляла 220 мг. Наиболее частыми нежелательными реакциями при данной дозе препарата были нежелательные реакции со стороны кожи, дисфония, диарея, воспаление слизистых оболочек, сухость слизистой оболочки полости рта, снижение аппетита, повышение АД, общая слабость.
Лечение: специфический антидот неизвестен. В случае передозировки прием регорафениба следует прекратить и применять стандартную симптоматическую терапию. Пациент должен находиться под наблюдением врача до стабилизации состояния.
Способ применения и дозы
Внутрь. Рекомендуемая суточная доза регорафениба составляет 160 мг 1 раз в сутки в течение 3 нед. В последующую неделю (4-я нед от начала лечения) следует сделать перерыв. Период продолжительностью 4 нед от начала приема является одним курсом лечения.
Регорафениб принимают в одно и то же время после приема пищи, содержащей низкое (<30%) количество жира (см. «Фармакология»).
Коррекция дозы. Индивидуальная переносимость и безопасность лечения может потребовать временного прекращения терапии и/или уменьшения дозы регорафениба. Коррекция дозы на каждом этапе ее снижения составляет 40 мг. Наименьшая рекомендуемая доза регорафениба составляет 80 мг/сут. Максимальная суточная доза — 160 мг.
Меры предосторожности
Действие на печень
У пациентов, получавших лечение регорафенибом, часто регистрировались отклонения значений биохимических показателей функции печени (AЛT, ACT, билирубин). У небольшой части пациентов отмечались тяжелые нарушения показателей функции печени (3–4-я степени тяжести) и клинически выраженные нарушения функции печени (в т.ч. с летальным исходом) (см. «Побочные действия»).
До начала лечения регорафенибом рекомендуется определить показатели функции печени (ACT, АЛТ, билирубин). На протяжении первых 2 мес терапии следует проводить контроль функции печени по меньшей мере каждые 2 нед, затем не реже 1 раза в месяц, а также согласно клиническим показателям.
Поскольку регорафениб является ингибитором UGT1A1, у пациентов с синдромом Жильбера возможно появление слабовыраженной непрямой (неконъюгированной) гипербилирубинемии (см. «Взаимодействие»).
Если у больных, получающих лечение регорафенибом, отмечается ухудшение показателей функции печени, связанное с терапией (в отсутствие явной альтернативной причины, например механической желтухи или прогрессирования основного заболевания), врачу следует изменить дозу препарата и наблюдать за состоянием пациента.
RxList.com
Гепатотоксичность
Во всех клинических испытаниях применения регорафениба с участием 1200 пациентов зарегистрированы эпизоды тяжелого лекарственноиндуцированного поражения печени с летальным исходом (0,3%). Имевшиеся результаты биопсии печени показали некроз гепатоцитов с инфильтрацией лимфоцитами. В исследовании 1 печеночная недостаточность со смертельным исходом наблюдалась у 1,6% пациентов, принимавших регорафениб, и у 0,4% пациентов группы плацебо; у всех больных с печеночной недостаточностью были метастазы в печени. В исследовании 2 печеночная недостаточность с летальным исходом была зарегистрирована у 0,8% пациентов, принимавших регорафениб (см. «Побочные действия»).
Необходимо исследовать показатели функции печени (АЛТ, АСТ, билирубин) перед началом лечения регорафенибом и контролировать их по крайней мере каждые 2 нед в течение первых 2 мес лечения. После этого проводить обследование пациента ежемесячно или чаще (по клиническим показаниям). При нарушении функии печени необходимо проводить еженедельный мониторинг значений АЛТ, АСТ и билирубина пока они не будут ниже уровня 3-кратной ВГН или соответствовать значениям до начала лечения.
В зависимости от степени тяжести и стойкости процесса лекарственной гепатотоксичности, проявляющейся в виде повышения значений печеночных проб и развития гепатоцеллюлярного некроза, необходимо либо временно поддерживать выбранное дозирование регорафениба, либо уменьшать дозу или даже прекращать проведение лечения.
RxList.com (обновление 2021 г.)
Гепатотоксичность
В ходе клинических испытаний у пациентов, получавших регорафениб, наблюдалось тяжелое лекарственное поражение печени с летальным исходом. В большинстве случаев нарушение функции печени возникало в течение первых 2 мес терапии и характеризовалось гепатоцеллюлярным характером повреждения.
В исследовании RESORCE не было увеличения частоты летальной печеночной недостаточности по сравнению с плацебо.
Инфекции
Регорафениб вызывал повышение риска инфекций. Общая частота инфекции (степени 1–5) была выше (32% против 17%) у 1142 пациентов, получавших регорафениб, по сравнению с контрольной группой в рандомизированных плацебо-контролируемых исследованиях. Частота инфекций 3-й степени или выше в группе больных, принимавших регорафениб, была 9%. Наиболее частыми инфекциями являлись инфекции мочевыводящих путей (5,7%), назофарингит (4%), инфекции кожи и слизистых оболочек и системные грибковые инфекции (3,3%), пневмония (2,6%). Летальные исходы, вызванные инфекцией, чаще возникали у пациентов, получавших регорафениб (1%), чем у пациентов, получавших плацебо (0,3%); наиболее частыми фатальными инфекциями были респираторные (0,6% у пациентов, получавших регорафениб, против 0,2% у пациентов, получавших плацебо).
Не следует принимать регорафениб при инфекциях 3-й или 4-й степени или при обострении инфекции любой степени. Можно возобновить лечение регорафенибом в той же дозе после разрешения инфекции.
Пациенты с опухолями и мутациями в гене KRAS
У пациентов с мутациями в гене KRAS наблюдалось значительное улучшение показателей выживаемости без прогрессирования и было зарегистрировано численно более слабое воздействие на общую выживаемость (см. «Фармакодинамика»). Принимая во внимание значительную токсичность, связанную с терапией, врачам рекомендуют внимательно оценивать пользу и риски при назначении регорафениба пациентам с опухолями, имеющими мутации в гене KRAS.
Кровотечение
У больных, получавших регорафениб, было зарегистрировано повышение частоты эпизодов кровотечений, в некоторых случаях с летальным исходом (см. «Побочные действия»). При наличии факторов риска кровотечения, а также при совместном назначении с антикоагулянтами (например варфарин, фенпрокумон) или другими ЛС, повышающими риск кровотечения, следует контролировать показатели коагулограммы и общего анализа крови. При появлении тяжелого кровотечения, требующего экстренного медицинского вмешательства, следует рассмотреть вопрос о прекращении лечения регорафенибом.
RxList.com (2016 г.)
Кровотечение
Применение регорафениба приводило к увеличению частоты случаев кровотечения. Общая частота возникновения кровотечения (1–5-й степени тяжести) составляла 21 и 11% больных, принимавших регорафениб, по сравнению с 8 и 3% пациентов, принивавших плацебо, в клинических исследованиях 1 и 2. Случаи кровоизлияния с летальным исходом зарегестрированы у 4 (0,6%) из 632 больных, принимавших регорафениб в обоих исследованиях, с локализацией в ЖКТ, дыхательной и мочеполовой системах.
Необходимо прервать лечение регорафенибом у больных с тяжелым или угрожающим жизни кровотечением и чаще определять уровни МНО у пациентов, принимающих варфарин.
RxList.com (обновление 2021 г.)
Кровотечение
Регорафениб вызывал повышенную частоту кровотечений. Общая частота возникновения кровотечения (1–5-й степени тяжести) составила 18,2% у 1142 пациентов, получавших регорафениб, и 9,5% у пациентов, получавших плацебо, в рандомизированных плацебо-контролируемых исследованиях. Частота кровотечения 3-й степени или выше у пациентов, получавших регорафениб, составила 3%. Частота смертельных геморрагических событий составила 0,7% с локализацией в ЦНС, ЖКТ, дыхательной и мочеполовой системах.
Ишемия миокарда и инфаркт миокарда
При приеме регорафениба отмечалось увеличение частоты случаев ишемии миокарда и инфаркта миокарда (см. «Побочные действия»).
Пациенты с нестабильной стенокардией или появлением стенокардии (в течение 3 мес до начала терапии регорафенибом), недавним инфарктом миокарда (в период 6 мес до начала терапии регорафенибом) и пациенты с сердечной недостаточностью 2-го класса и выше по классификации Нью-Йоркской кардиологической ассоциации были исключены из клинических исследований.
У пациентов с ИБС необходимо отслеживать клинические признаки и симптомы ишемии миокарда. При возникновении ишемии и/или инфаркта миокарда следует прекратить терапию регорафенибом до нормализации состояния. При принятии решения о возобновлении терапии врач должен оценивать соотношение пользы от приема ЛС с потенциальным риском у каждого отдельного пациента. Если клинические проявления ишемии сохраняются, возобновлять терапию не следует.
RxList.com (2016 г.)
Ишемия и инфаркт миокарда
Применение регорафениба увеличило частоту возникновения ишемии и инфаркта миокарда в исследовании 1 (1,2 против 0,4%) (см. «Побочные действия»). Необходимо прекратить лечение регорафенибом больных, у которые происходит обострение ИБС или развивается ишемия и/или инфаркт миокарда. Возобновление терапии регорафенибом после разрешения случая острой ишемии миокарда возможно, только если потенциальная польза лечения превышает риск дальнейшего развития ИБС.
RxList.com (обновление 2021 г.)
Ишемия и инфаркт миокарда
В рандомизированных плацебо-контролируемых исследованиях регорафениб увеличивал частоту возникновения ишемии и инфаркта миокарда (0,9 против 0,2%) (см. «Побочные действия»).
Синдром обратимой задней энцефалопатии
У пациентов, получавших регорафениб, были зарегистрированы случаи развития обратимой задней энцефалопатии (см. «Побочные действия»). Обратимая задняя энцефалопатия проявлялась в виде судорог, головной боли, изменения сознания, нарушения зрения или корковой слепоты, иногда в сочетании с артериальной гипертензией. Для подтверждения диагноза пациентам следует провести томографию головного мозга. В случае развития обратимой задней энцефалопатии следует прекратить лечение регорафенибом, проводить контроль АД и поддерживающую терапию.
RxList.com (2016 г.)
Синдром обратимой задней лейкоэнцефалопатии
Синдром обратимой задней лейкоэнцефалопатии, синдром подкоркового вазогенного отека, диагностируемый по характерным признакам с помощью МРТ, наблюдался у 1 из 1200 пациентов, принимавших регорафениб во всех клинических испытаниях. Необходимо провести диагностику этого синдрома у всех пациентов с эпилептическими припадками, головной болью, нарушением зрения, спутанностью сознания или измененной психической функцией и прервать терапию регорафенибом при развитии синдрома обратимой задней лейкоэнцефалопатии.
RxList.com (обновление 2021 г.)
Синдром обратимой задней лейкоэнцефалопатии
Синдром обратимой задней лейкоэнцефалопатии, синдром подкоркового вазогенного отека, диагностируемый по характерным признакам с помощью МРТ, наблюдался у 1 из 4800 пациентов, принимавших регорафениб во всех клинических испытаниях.
Прободение и свищ ЖКТ
У пациентов, получавших регорафениб, были зарегистрированы случаи прободения ЖКТ и образования свища ЖКТ (см. «Побочные действия»). Эти события были связаны с опухолями в брюшной полости. В случае прободения ЖКТ или образования свища терапию регорафенибом следует прекратить.
RxList.com (2016 г.)
Прободение или свищ ЖКТ
Прободение или свищ ЖКТ наблюдались при приеме регорафениба в 0,6% случаев, включая 4 летальных исхода, у 1200 пациентов во всех клинических испытаниях. В исследовании 2 у 2,1% (4 из 188) больных, принимавших регорафениб в течение слепой или открытой части этого исследования, было зафиксировано развитие свища или прободения ЖКТ; 2 случая прободения ЖКТ были с летальным исходом. Необходимо прервать терапию регорафенибом при развитии прободения или свища ЖКТ.
RxList.com (обновление 2021 г.)
Прободение или свищ ЖКТ
Прободение или свищ ЖКТ наблюдались при приеме регорафениба в монотерапии у 0,6% из 4518 пациентов, включая 8 летальных исходов, во всех клинических испытаниях.
В рандомизированных плацебо-контролируемых исследованиях развитие желудочно-кишечного свища было зафиксировано у 0,8% пациентов, получавших регорафениб, и у 0,2% пациентов в группе плацебо.
Повышение АД
На фоне терапии регорафенибом было зарегистрировано увеличение частоты повышения АД (см. «Побочные действия»). Перед началом и во время лечения регорафенибом следует регулярно контролировать АД и корректировать его повышение в соответствии с принятыми стандартами лечения. В случаях развития тяжелой или стойкой артериальной гипертензии, устойчивой к проводимой адекватной антигипертензивной терапии, врач должен временно прервать терапию и/или снизить дозу регорафениба (см. «Способ применения и дозы»). В случае развития гипертонического криза лечение регорафенибом следует отменить.
RxList.com (2016 г.)
Гипертензия
Применение регорафениба вызвало повышение количества случаев возникновения гипертензии (30 против 8% в исследовании 1 и 59% по сравнению с 27% в исследовании 2) (см. «Побочные действия»). Гипертонический криз был зарегистрирован в 0,25% случаев из 1200 больных, принимавших регорафениб во всех клинических испытаниях. Возникновение гипертензии во время первого цикла лечения зафиксировано у большинства пациентов с выявленной ранее гипертензией (72% в исследовании 1 и исследовании 2).
Нельзя назначать регорафениб, пока АД адекватно не контролируется. Необходимо еженедельно мониторировать АД в течение первых 6 нед лечения, а затем — в течение каждого цикла или чаще (по клиническим показаниям). Необходимо временно воздержаться от терапии регорафенибом или полностью прекратить лечение при тяжелой или неконтролируемой артериальной гипертензии.
RxList.com (обновление 2021 г.)
Гипертензия
В рандомизированных плацебо-контролируемых исследованиях гипертонический криз был зарегистрирован у 0,2% пациентов в группах регорафениба и ни у одного из пациентов в группах плацебо. Регорафениб вызвал увеличение случаев возникновения гипертензии (30 против 8% в исследовании CORRECT, 59 против 27% в исследовании GRID и 31 против 6% в исследовании RESORCE) (см. «Побочные действия»). Развитие гипертензии наблюдалось во время первого цикла лечения у большинства пациентов, у которых развилась артериальная гипертензия (67% в рандомизированных плацебо-контролируемых исследованиях).
Нарушение заживления ран
В случае проведения обширных хирургических вмешательств рекомендуется временное прекращение терапии регорафенибом, поскольку ЛС, обладающие антиангиогенными свойствами, могут подавлять или ухудшать заживление ран. Решение о возобновлении терапии после хирургических вмешательств должно основываться на клинической оценке адекватности заживления раны.
RxList.com (2016 г.)
Заживление ран
Исследования о влиянии регорафениба на заживление ран формально не проводились. Так как ингибиторы активности рецептора сосудистого эндотелиального фактора роста (VEGFR), такие как регорафениб, могут ухудшать заживление ран, лечение регорафенибом должно быть прекращено по крайней мере за 2 нед до запланированной операции. Решение о возобновлении терапии регорафенибом после операции должно основываться на клинической оценке адекватного заживления ран. Лечение регорафенибом должно быть прекращено при наличии у больного зияющей раны.
RxList.com (обновление 2021 г.)
Риск нарушения заживления ран
Осложнения, связанные с нарушением заживления ран, могут возникать у пациентов, принимающих ЛС, ингибирующие сигнальный путь VEGF. Следовательно, регорафениб может отрицательно повлиять на заживление ран.
Необходимо воздержаться от приема регорафениба в течение как минимум 2 нед до плановой операции, а также не применять его в течение как минимум 2 нед после серьезной операции и до адекватного заживления ран. Безопасность возобновления приема регорафениба после разрешения осложнений при заживлении ран не установлена.
Кожная токсичность
Наиболее частыми нежелательными реакциями при приеме регорафениба были ладонно-подошвенная эритродизестезия (ЛПЭ) и сыпь (см. «Побочные действия»). С целью профилактики развития ЛПЭ следует контролировать образование мозолей и использовать специальные вкладыши для обуви и перчатки для предотвращения давления на подошвы и ладони. Для лечения ЛПЭ можно использовать кератолитические кремы (например кремы на основе мочевины, салициловой кислоты или альфа-гидроксильной кислоты, которые следует наносить только на пораженные участки кожи) и увлажняющие кремы в обильном количестве для облегчения симптомов. При необходимости временно прекращают лечение и/или снижают дозу регорафениба или, в тяжелых или повторяющихся случаях кожных реакций, терапию регорафенибом прекращают.
RxList.com (2016 г.)
Кожная токсичность
Применение регорафениба приводило к повышению частоты возникновения побочных реакций с поражением кожи и подкожных тканей (72% по сравнению 24% в исследовании 1 и 78% по сравнению с 24% в исследовании 2), в т.ч. ЛПЭ и сыпи тяжелой степени, что требует снижения принимаемой дозы этого ЛС.
Общая частота встречаемости ЛПЭ была выше у больных, принимавших регорафениб (45 против 7% в исследовании 1 и 67% по сравнению с 12% в исследовании 2), чем у пациентов, получавших плацебо. В большинстве случаев ЛПЭ у больных, принимавших регорафениб, регистрировалась во время первого цикла лечения (69 и 71% пациентов, у которых развивалась ЛПЭ в исследовании 1 и исследовании 2 соответственно). Частота встречаемости ЛПЭ 3-й степени тяжести (17 против 0% в исследовании 1 и 22% по сравнению с 0% в исследовании 2) и сыпи 3-й степени тяжести (6% по сравнению с <1% в исследовании 1 и 7% по сравнению с 0% в исследовании 2), серьезные неблагоприятные реакции в виде мультиформной эритемы (0,2 против 0% в исследовании 1) и синдрома Стивенса-Джонсона (0,2 против 0% в исследовании 1) была выше у больных, принимавших регорафениб (см. «Побочные действия»).
Токсический эпидермальный некролиз был выявлен в 0,17% случаев у 1200 больных, принимавших регорафениб во всех клинических испытаниях.
Необходимо снижать дозировку либо прекратить лечение регорафенибом, в зависимости от тяжести и стойкости дерматологической токсичности, и назначить дополнительную терапию для облегчения ее симптомов.
RxList.com (обновление 2021 г.)
Кожная токсичность
В рандомизированных плацебо-контролируемых исследованиях побочные кожные реакции возникали у 71,9% пациентов в группе регорафениба и у 25,5% пациентов в группе плацебо, включая ЛПР, также известную как ЛПЭ, и сыпь тяжелой степени, что требует снижения принимаемой дозы.
В рандомизированных плацебо-контролируемых исследованиях с участием 1142 пациентов общая частота возникновения ЛПР была выше у больных, принимавших регорафениб (53%), чем у пациентов, получавших плацебо (8%). В большинстве случаев ЛПР у больных, принимавших регорафениб, регистрировалась во время первого цикла лечения. Частота встречаемости ЛПР 3-й степени тяжести (16 против <1%), сыпи 3-й степени тяжести (3% по сравнению с <1%), серьезных неблагоприятных реакций в виде многоформной эритемы (<0,1 против 0%) и синдрома Стивенса-Джонсона (<0,1 против 0%) была выше у больных, принимавших регорафениб (см. «Побочные действия»).
Во всех исследованиях более высокая частота ЛПР наблюдалась у азиатских пациентов, получавших регорафениб (все степени — 72%, 3-я степень — 18%).
Токсический эпидермальный некролиз был выявлен в 0,02% из 4518 больных, принимавших регорафениб в монотерапии во всех клинических испытаниях.
Отклонения значений лабораторных показателей
При применении регорафениба было зарегистрировано повышение частоты электролитных нарушений (включая гипофосфатемию, гипокальциемию, гипонатриемию и гипокалиемию) и нарушений метаболизма (включая увеличение концентрации ТТГ и активности амилазы). Отклонения от нормы обычно носили легкий или умеренный характер и не сопровождались клиническими проявлениями. В случае возникновения электролитных нарушений или нарушений метаболизма коррекция дозы или прекращение терапии не требуется. Во время терапии регорафенибом рекомендуется контролировать биохимические и метаболические показатели. При необходимости назначают заместительную терапию в соответствии с принятыми стандартами лечения. В случаях устойчивых или рецидивирующих нарушений следует рассмотреть возможность временного прекращения лечения или снижения дозы, или полного прекращения терапии регорафенибом (см. «Способ применения и дозы»).
Системная токсичность
После повторного введения дозы у мышей, крыс и собак наблюдались нежелательные реакции со стороны ряда органов, прежде всего почек, печени, пищеварительного тракта, щитовидной железы, лимфатической/кроветворной системы, эндокринной системы, репродуктивной системы и кожи. В 26-недельном исследовании токсичности при введении повторных доз у крыс наблюдалось небольшое увеличение частоты случаев утолщения атриовентрикулярных клапанов сердца. Причиной данного явления может служить ускорение возрастного физиологического процесса. Данные реакции наблюдались при системных экспозициях, находящихся в диапазоне или ниже диапазона предполагаемой экспозиции у человека (основано на сравнении AUC).
Изменения зубов и костей, а также нежелательные явления в репродуктивной системе были наиболее выражены у молодых и растущих животных, в т.ч. у молодых крыс, что указывает на потенциальный риск для детей и подростков.
Тератогенность и эмбриотоксичность
Специальные исследования влияния регорафениба на фертильность не проводились. Следует учитывать способность регорафениба оказывать неблагоприятное воздействие на репродуктивную систему мужчин и женщин. В исследовании на крысах и собаках после многократного применения регорафениба при экспозициях ниже предполагаемой экспозиции у человека (при сравнении AUC) наблюдались морфологические изменения в яичках, яичниках и матке. Наблюдаемые изменения были обратимы только частично. В исследовании на кроликах при экспозиции ниже предполагаемой экспозиции у человека наблюдалась эмбриотоксичность (при сравнении AUC). В основном были обнаружены нарушения формирования мочевыделительной системы, сердца и крупных сосудов, костей.
RxList.com (обновление 2021 г.)
Эмбриофетальная токсичность
На основании исследований на животных и механизма действия, регорафениб может нанести вред плоду при применении беременной женщиной. Нет доступных данных о применении регорафениба у беременных. Регорафениб оказывал эмбриолетальное и тератогенное действие у крыс и кроликов при более низких экспозициях, чем экспозиции у человека при применении в рекомендованной дозе, и вызывал возникновение пороков развития сердечно-сосудистой, мочеполовой и скелетно-мышечной систем с повышенной частотой. Необходимо сообщить беременным женщинам о потенциальном риске для плода.
Женщинам с репродуктивным потенциалом и их партнерам-мужчинам следует использовать эффективную контрацепцию во время лечения регорафенибом и в течение 2 мес после последней дозы.
Особые группы пациентов
Нарушение функции печени. Печень имеет большое значение в выведении регорафениба. Не было выявлено клинически значимых различий в воздействии регорафениба у пациентов со средним (класс А по классификации Чайлд-Пью) и умеренным (класс В по классификации Чайлд-Пью) нарушением функции печени в сравнении с пациентами с нормальной функцией печени. Пациентам со средним и умеренным нарушением функции печени коррекция дозы не требуется. Рекомендуется проводить мониторинг функции печени на фоне терапии регорафенибом у данной категории пациентов (см. «Фармакология», «Меры предосторожности»). Не рекомендуется лечение регорафенибом пациентов с тяжелым нарушением функции печени (класс С по классификации Чайлд-Пью), поскольку применение препарата у данной категории пациентов не изучено (возможно увеличение экспозиции этого ЛС).
RxList.com (обновление 2021 г.)
Печеночная недостаточность. Коррекция дозы не рекомендуется у пациентов с легкой (общий билирубин ≤ВГН и AСT >ВГН или общий билирубин >ВГН до ≤1,5 ВГН) или умеренной (общий билирубин от >1,5 до ≤3 ВГН и любая AСT) печеночной недостаточностью. Рекомендуется тщательный мониторинг пациентов с нарушением функции печени на предмет развития побочных реакций.
Регорафениб не рекомендуется применять пациентам с тяжелой печеночной недостаточностью (общий билирубин >3 ВГН), т.к. он не исследовался у этой популяции.
Нарушение функции почек. В клинических исследованиях не отмечалось значимых различий безопасности и эффективности регорафениба у пациентов с легкой степенью почечной недостаточности (расчетная СКФ 60–89 мл/мин/1,73 м2) в сравнении с пациентами с нормальной функцией почек. Ограниченные данные по фармакокинетике не выявили различий в экспозиции у пациентов со средней степенью почечной недостаточности (расчетная СКФ 30–59 мл/мин/1,73 м2). Пациентам с легкой и средней степенью почечной недостаточности снижение дозы препарата не требуется (см. «Фармакология»). Применение регорафениба у пациентов с тяжелой степенью почечной недостаточности (расчетная СКФ <30 мл/мин/1,73 м2) не изучено.
RxList.com (обновление 2021 г.)
Почечная недостаточность. Коррекция дозы не рекомендуется у пациентов с почечной недостаточностью. Фармакокинетика регорафениба не изучалась у пациентов, находящихся на диализе, и нет рекомендованной дозы для этой популяции пациентов (см. «Фармакокинетика»).
Дети. Безопасность и эффективность назначения регорафениба у детей и подростков до 18 лет не установлена.
RxList.com (обновление 2021 г.)
Данные у животных
В 28-дневных исследованиях повторных доз у крыс были выявлены дозозависимые изменения дентина и ангиэктазии. Эти результаты были получены при дозах регорафениба менее 4 мг/кг (примерно 25% AUC человека при рекомендованной дозе). В 13-недельных исследованиях повторных доз у собак были аналогичные результаты изменения дентина при дозах менее 20 мг/кг (примерно 43% AUC у человека при рекомендованной дозе). Применение регорафениба у этих животных также приводило к стойкому росту и утолщению эпифизарной ростовой пластинки бедренной кости.
Пожилой возраст. В клинических исследованиях не отмечалось значимых различий безопасности и эффективности регорафениба у пожилых (65 лет и старше) пациентов в сравнении с более молодыми пациентами. Пациентам пожилого возраста коррекция дозы не требуется (см. «Фармакология»).
RxList.com (обновление 2021 г.)
Применение в гериатрии. Из 1142 пациентов, получавших регорафениб и включенных в рандомизированные плацебо-контролируемые исследования, 40% были в возрасте 65 лет и старше, а 10% — 75 лет и старше. Каких-либо общих различий в эффективности у этих пациентов и более молодых пациентов не наблюдалось. В плацебо-контролируемых исследованиях среди пациентов в возрасте 65 лет и старше, принимавших регорафениб, отмечалось повышение частоты гипертензии 3-й степени (18% против 9%) по сравнению с более молодыми пациентами. Кроме того, один эпизод артериальной гипертензии 4-й степени был зарегистрирован в возрастной группе 65 лет и старше и ни одного случая в более молодой возрастной группе.
Половая принадлежность. В клинических исследованиях не отмечалось значимых различий безопасности и эффективности регорафениба у пациентов мужского и женского пола. Коррекция дозы регорафениба в зависимости от пола пациента не требуется (см. «Фармакология»).
Этническая принадлежность. В клинических исследованиях не отмечалось значимых различий безопасности и эффективности регорафениба у пациентов разных этнических групп. Корректировка дозы этого ЛС в зависимости от этнической принадлежности пациента не требуется (см. «Фармакология»).
RxList.com (обновление 2021 г.)
Раса. Основываясь на объединенных данных трех плацебо-контролируемых исследований (CORRECT, GRID и CONCUR), у азиатов, получавших регорафениб, по сравнению с белокожими наблюдалась более высокая частота ЛПР и аномалий тестов функции печени. Коррекция начальной дозы в зависимости от расы не требуется.
Влияние на способность управлять транспортными средствами и механизмами. Специальные исследования не проводились, однако при возникновении нежелательных явлений, которые могут влиять на указанные способности, рекомендуется избегать управления транспортными средствами и занятий другими потенциально опасными видами деятельности, требующими повышенной концентрации внимания и быстроты психомоторных реакций (до исчезновения данных симптомов).
Источники информации
www.grls.rosminzdrav.ru, 2016, и www.rxlist.com, 2016, www.rxlist.com, 2021.
Торговые названия с действующим веществом Регорафениб
| Торговое название | Цена за упаковку, руб. |
|---|---|
| Стиварга® |
от 176800.00 до 181030.00 |
Rec.INN
зарегистрированное ВОЗ
Лекарственное взаимодействие
Входит в состав препаратов:
список
Фармакологическое действие
Противоопухолевое средство. Регорафениб является ингибитором многочисленных протеинкиназ, включая киназы, участвующие в ангиогенезе опухоли (VEGFRl,-2,-3, TIE2), онкогенезе (KIT, RET, RAF-1, BRAF, BRAFv600E), метастазировании (VEGFR3, PDGFR, FGFR), а также в противоопухолевом иммунном ответе (CSF1R). В частности, регорафениб ингибирует мутантную киназу KIT, ключевой онкогенный фактор в развитии стромальных опухолей ЖКТ. Благодаря этому регорафениб блокирует пролиферацию опухолевых клеток. В доклинических исследованиях было показано, что регорафениб оказывает выраженное противоопухолевое действие на широком спектре опухолевых моделей, включая колоректальный рак, гастроинтестинальные стромальные опухоли и печеночно-клеточный рак. Эффект регорафениба возможно связан с его антиангиогенным и антипролиферативным воздействием. Кроме того, регорафениб снижает уровень макрофагов, ассоциированных с опухолью, и показывает антиметастатическое действие in vivo.Основные метаболиты препарата в организме человека (М-2 и М-5) по своей эффективности на моделях in vitro и in vivo сопоставимы с регорафенибом.
Фармакокинетика
После приема внутрь средняя относительная биодоступность регорафениба составляет 69-83%.
Среднее значение Cmax в плазме крови составляет около 2.5 мг/л приблизительно через 3-4 ч после однократной пероральной дозы регорафениба 160 мг. Наибольшая концентрация регорафениба и его основных фармакологически активных метаболитов М-2 (N-оксид) и М-5 (N-оксид и N-дезметил) достигается после приема завтрака с низким содержанием жиров по сравнению с приемом после завтрака с высоким содержанием жиров или приемом натощак. По сравнению с приемом натощак экспозиция регорафениба увеличивается на 48% при приеме после завтрака с высоким содержанием жиров и на 36% при приеме после завтрака с низким содержанием жиров. По сравнению с приемом натощак, экспозиция метаболитов М-2 и М-5 выше при приеме регорафениба после завтрака с низким содержанием жиров и ниже при приеме после завтрака с высоким содержанием жира.
Кривая зависимости «концентрация-время» демонстрирует несколько пиков как для регорафениба, так и для его основных циркулирующих метаболитов, в течение 24 ч после приема дозы, что связано с печеночно-кишечной рециркуляцией. Связывание регорафениба с белками плазмы крови in vitro высокая и составляет 99.5%.
Метаболизм регорафениба осуществляется главным образом в печени путем окисления, опосредованного изоферментом CYP3A4, а также путем глюкуронирования, опосредованного UGT1A9. В плазме выявляются 2 основных и 6 второстепенных метаболитов регорафениба. Основные циркулирующие в плазме крови метаболиты регорафениба М-2 (N-оксид) и М-5 (N-оксид и N-дезметил) обладают фармакологической активностью и в равновесном состоянии имеют концентрации, сходные с концентрацией регорафениба.
In vitro связывание М-2 и М-5 с белками крови выше, чем у регорафениба и составляет 99.8% и 99.95 соответственно. Метаболиты могут быть восстановлены и гидролизированы микрофлорой ЖКТ, при этом возможно повторное всасывание неконъюгированного активного вещества и его метаболитов (печеночно-кишечная рециркуляция).
T1/2 регорафениба и его метаболита М-2 из плазмы составляет от 20 до 30 ч после приема внутрь. Средний T1/2 метаболита М-5 составляет около 60 ч (40-100 ч).
Приблизительно 90% дозы регорафениба, меченого радиоактивным изотопом, выводится в течение 12 сут после его приема, при этом 71% выводится через кишечник (47% в виде неизмененного вещества и 24% в виде метаболитов) и около 19% — почками в виде глюкуронидов. В равновесном состоянии выведение глюкуронидов почками уменьшается и составляет меньше 10%. Исходное соединение, обнаруженное в каловых массах, может быть продуктом желудочно-кишечного расщепления глюкуронидов, или продуктом восстановления метаболита М-2 (N-оксида), или остатком неабсорбированного препарата.
Системное воздействие регорафениба при равновесной концентрации возрастает пропорционально дозе при дозе до 60 мг и менее пропорционально при дозах более 60 мг. Накопление активного вещества при равновесной концентрации приблизительно в 2 раза превышает концентрацию в плазме, которая соответствует T1/2 и частоте приема лекарственного средства.
Поскольку печень имеет большое значение в выведении регорафениба, у пациентов с тяжелыми нарушениями функции печени возможно усиление его действия.
У пациентов с легкой, средней или тяжелой степенью почечной недостаточности экспозиция регорафениба и его метаболитов М-2 и М-5 в равновесном состоянии являлась такой же, как у пациентов с нормальной функцией почек. У пациентов с тяжелой степенью почечной недостаточности экспозиция регорафениба в равновесном состоянии была такой же, как и у пациентов с нормальной функцией почек, в то время как экспозиция М-2 и М-5 уменьшалась приблизительно на 30%. У пациентов с терминальной степенью почечной недостаточности фармакокинетика регорафениба не изучена.
Показания активного вещества
РЕГОРАФЕНИБ
Метастатический колоректальный рак у пациентов, которым уже проводилась или не показана химиотерапия фторпиримидиновыми препаратами, терапия, направленная против сосудистого эндотелиального фактора роста (VEGF), и терапия, направленная против рецепторов эпидермального фактора роста (EGFR).
Неоперабельные или метастатические гастроинтестинальные стромальные опухоли у пациентов при прогрессировании на терапии иматинибом и сунитинибом или при непереносимости данного вида лечения.
Печеночно-клеточный рак у пациентов, которым уже проводилась терапия сорафенибом.
Режим дозирования
Для приема внутрь.
Рекомендуемая доза составляет 160 мг 1 раз/сут в течение 3 недель. В последующую неделю (4-я неделя от начала лечения) следует перерыв. Один курс лечения — это период 4 недели от начала приема.
Лечение продолжают до тех пор, пока сохраняется клиническая эффективность или до появления неприемлемого токсического действия.
Индивидуальная переносимость и безопасность лечения может потребовать временного прекращения терапии и/или уменьшения дозы.
Побочное действие
Со стороны системы кроветворения: очень часто — тромбоцитопения, анемия; часто — лейкопения.
Со стороны иммунной системы: нечасто — реакции повышенной чувствительности.
Со стороны сердечно-сосудистой системы: очень часто — кровотечения, повышение АД; нечасто — инфаркт миокарда, ишемия миокарда, гипертонический криз.
Со стороны дыхательной системы: очень часто — дисфония.
Со стороны кожи и подкожных тканей: очень часто — ладонно-подошвенная эритродизестезия, кожная сыпь; часто — алопеция, сухость кожи, эксфолиативный дерматит; нечасто — поражение ногтей, многоформная эритема; редко — синдром Стивенса-Джонсона, токсический эпидермальный некролиз.
Со стороны пищеварительной системы: очень часто — диарея, стоматит, рвота, тошнота; часто — нарушение вкуса, сухость слизистой оболочки полости рта, гастроэзофагеальный рефлюкс, гастроэнтерит; нечасто — прободение ЖКТ, свищ ЖКТ, панкреатит.
Со стороны печени и желчевыводящих путей: очень часто — гипербилирубинемия, повышение активности трансаминаз; нечасто — тяжелое нарушение функции печени.
Со стороны нервной системы: часто — головная боль, тремор; редко — синдром задней обратимой энцефалопатии.
Со стороны костно-мышечной системы: часто — мышечные спазмы.
Со стороны мочевыделительной системы: часто — протеинурия.
Со стороны эндокринной системы: часто — гипотиреоз.
Со стороны обмена веществ: очень часто — снижение аппетита и потребления пищи, снижение массы тела; часто — гипокалиемия, гипофосфатемия, гипокальциемия, гипонатриемия, гипомагниемия, гиперурикемия, дегидратация.
Лабораторные данные: часто — увеличение активности амилазы и липазы, отклонение от нормального значения МНО.
Доброкачественные, злокачественные и неуточненные новообразования (включая кисты и полипы): редко — кератоакантома/плоскоклеточный рак кожи.
Инфекционные и паразитарные заболевания: часто — инфекции мочевыводящих путей, назофарингит, кандидоз кожи и слизистых, системный микоз, пневмония.
Общие реакции: очень часто — астения/общая слабость, боль различной локализации, повышение температуры тела, воспаление слизистых оболочек.
Противопоказания к применению
Тяжелая степень печеночной недостаточности (класс С по классификации Чайлд-Пью); терминальная степень почечной недостаточности; совместное применение с сильными ингибиторами и индукторами CYP3A4; возраст до 18 лет; беременность и период лактации (грудного вскармливания); повышенная чувствительность к регорафенибу.
Применение при беременности и кормлении грудью
Противопоказано применение при беременности и в период лактации (грудного вскармливания).
Применение при нарушениях функции печени
Противопоказано применение при тяжелая степени печеночной недостаточности (класс С по классификации Чайлд-Пью).
Применение при нарушениях функции почек
Противопоказано применение при терминальной степени почечной недостаточности.
Применение у детей
Препарат противопоказан для применения у детей и подростков в возрасте до 18 лет
Особые указания
С осторожностью применять при нарушениях функции печени легкой и средней степени тяжести; при наличии факторов риска кровотечения, а также при совместном применении с антикоагулянтами и другими препаратами, повышающими риск кровотечений; при ИБС.
До начала лечения регорафенибом рекомендуется определить показатели функции печени (ACT, АЛТ, билирубин). На протяжении первых двух месяцев терапии следует проводить контроль функции печени по меньшей мере каждые 2 недели, затем не реже 1 раза в месяц, а также согласно клиническим показателям.
Поскольку регорафениб является ингибитором UGT1A1, у пациентов с синдромом Жильбера возможно появление слабовыраженной непрямой (неконъюгированной) гипербилирубинемии.
Если у пациентов, получающих лечение регорафенибом, отмечается ухудшение показателей функции печени, связанное с терапией (в отсутствие явной альтернативной причины, например, механической желтухи или прогрессирования основного заболевания), врачу следует изменить дозу регорафениба и наблюдать за состоянием пациента.
При применении у пациентов с легким и умеренным нарушением функции печени следует проводить тщательный контроль за состоянием пациента. Противопоказано применение у пациентов с тяжелым нарушением функции печени (класс С по классификации Чайлд-Пью), поскольку регорафениб не изучен у данной категории пациентов (возможно увеличение экспозиции).
В случаях ухудшения состояния на фоне прогрессирования инфекции следует рассмотреть вопрос о приостановке лечения регорафенибом.
При наличии факторов риска кровотечения, а также при совместном назначении с антикоагулянтами (например, варфарин, фенпрокумон) или другими лекарственными препаратами, повышающими риск кровотечения, следует контролировать показатели коагулограммы и общего анализа крови. Перед началом лечения регорафенибом у пациентов с циррозом печени следует проводить обследование и последующее лечение варикозно расширенных вен пищевода в соответствии со стандартным подходом. При появлении тяжелого кровотечения, требующего экстренного медицинского вмешательства, следует рассмотреть вопрос о прекращении лечения регорафенибом.
В случае прободения ЖКТ или образования свища терапию регорафенибом следует прекратить.
У пациентов с ИБС необходимо отслеживать клинические признаки и симптомы ишемии миокарда. При возникновении ишемии и/или инфаркта миокарда следует прекратить терапию регорафенибом до нормализации состояния. При принятии решения о возобновлении терапии врач должен оценивать соотношение пользы от приема регорафениба с потенциальным риском у каждого отдельного пациента. Если клинические проявления ишемии сохраняются, возобновлять терапию не следует.
В случае развития обратимой задней энцефалопатии следует прекратить лечение регорафенибом, проводить контроль АД и поддерживающую терапию.
Перед началом и во время лечения регорафенибом следует регулярно контролировать АД и корректировать его повышение в соответствии с принятыми стандартами лечения. В случаях развития тяжелой или стойкой артериальной гипертензии, устойчивой к проводимой адекватной антигипертензивной терапии, врач должен временно прервать терапию и/или снизить дозу регорафениба. В случае развития гипертонического криза лечение регорафенибом следует отменить.
В случае проведения обширных хирургических вмешательств рекомендуется временное прекращение терапии регорафенибом, поскольку лекарственные препараты, обладающие антиангиогенными свойствами, могут подавлять или ухудшать заживление ран. Решение о возобновлении терапии после хирургических вмешательств должно основываться на клинической оценке адекватности заживления раны.
С целью профилактики развития ладонно-подошвенной эритродизестезии следует контролировать образование мозолей и использовать специальные вкладыши для обуви и перчатки для предотвращения давления на подошвы и ладони. Для лечения ладонно-подошвенной эритродизестезии можно использовать кератолитические кремы (например, кремы на основе мочевины, салициловой кислоты или альфа-гидроксильной кислоты, которые следует наносить только на пораженные участки кожи) и увлажняющие кремы в обильном количестве для облегчения симптомов. При необходимости временно прекращают лечение и/или снижают дозу регорафениба или в тяжелых или повторяющихся случаях кожных реакций терапию регорафенибом прекращают.
Следует контролировать биохимические и метаболических показатели. При необходимости назначают заместительную терапию в соответствии с принятыми стандартами лечения. В случаях устойчивых или рецидивирующих нарушений следует рассмотреть возможность временного прекращения лечения, снижения дозы или полного прекращения терапии регорафенибом.
Влияние на способность к управлению транспортными средствами и механизмами
При возникновении нежелательных явлений, которые могут влиять на указанные способности, рекомендуется избегать управления транспортными средствами и занятий другими потенциально опасными видами деятельности, требующими повышенной концентрации внимания и быстроты психомоторных реакций (до исчезновения данных симптомов).
Лекарственное взаимодействие
Применение кетоконазола (400 мг в течение 18 дней), сильного ингибитора изофермента CYP3A4, в сочетании с однократным приемом регорафениба (160 мг на пятый день) приводило к увеличению среднего воздействия (AUC) регорафениба приблизительно на 33% и снижению среднего воздействия его активных метаболитов, М-2 (N-оксид) и М-5 (N-оксид и N-дезметил) приблизительно на 90%. Не рекомендуется применять регорафениб совместно с сильными ингибиторами CYP3A4 (например, кларитромицин, грейпфрутовый сок, итраконазол, кетоконазол, позаконазол, телитромицин и вориконазол), поскольку их влияние на воздействие регорафениба в стабильном состоянии и его метаболитов (М-2 и М-5) не изучалось.
Применение рифампина (600 мг в течение 9 дней), сильного индуктора CYP3A4, в сочетании с однократным приемом регорафениба (160 мг на седьмой день) приводило к снижению среднего воздействия (AUC) регорафениба приблизительно на 50%, увеличению среднего воздействия активного метаболита М-5 в 3-4 раза, при этом не отмечалось изменения экспозиции активного метаболита М-2. Другие сильные ингибиторы CYP3A4 (например, фенитоин, карбамазепин, фенобарбитал и препараты, содержащие зверобой продырявленный) могут увеличивать метаболизм регорафениба. Не рекомендуется применять регорафениб совместно с сильными индукторами CYP3A4 или подбирать лекарственные средства, которые не влияют на CYP3A4 или индуцируют его в минимальной степени.
In vitro было показано, что регорафениб, также как и его активный метаболит М-2, подавляет глюкуронизацию под действием UGT1A1 и UGT1А9, в то время как метаболит М-5 подавляет UGT1A1 только в концентрациях, которые достигаются в равновесном состоянии in vivo.
Применение регорафениба с последующим 5-дневным перерывом перед назначением иринотекана приводило к увеличению среднего воздействия (AUC) SN-38 , субстрата UGT1А1 и активного метаболита иринотекана приблизительно на 44%. Также отмечалось увеличение среднего воздействия (AUC) иринотекана приблизительно на 28%. Эти данные показывают, что комбинированное применение регорафениба может увеличивать системную экспозицию субстратов UGT1A1 и UGT1A9.
Применение регорафениба (160 мг в течение 14 дней) перед однократным приемом розувастатина (5 мг), который является субстратом BCRP, приводило к увеличению среднего воздействия (AUC) розувастатина в 3.8 раз и к увеличению его средней Cmax в плазме крови в 4.6 раза. Комбинированное применение с регорафенибом может повышать концентрацию в плазме других субстратов BCRP (например, метотрексат, флувастатин, аторвастатин). Рекомендуется контролировать состояние пациентов с целью выявления признаков и симптомов увеличения экспозиции к субстратам BCRP.
Исследования in vitro показывают, что метаболиты регорафениба М-2 и М-5 являются субстратами Р-гликопротеина и BCRP. Ингибиторы и стимуляторы BCRPи Р-гликопротеина могут препятствовать экспозиции М-2 и М-5. Клиническая значимость данных результатов исследований неизвестна.
In vitro было показано, что регорафениб является конкурентным ингибитором цитохромов CYP2C8, CYP2C9, CYP2B6 в концентрациях, которые достигаются in vivo в стабильном состоянии (Cmax в плазме 8.1 мкмоль). In vitro ингибирующее действие в отношении CYP3A4 и CYP2C19 менее выражено.
Комбинированное применение регорафениба с неомицином не оказывает влияние на экспозицию регорафениба, но может нарушить его печеночно-кишечную циркуляцию. Тем не менее при сравнении фармакологической активности регорафениба in vivo и in vitro было показано уменьшение экспозиции его активных метаболитов М-2 и М-5 приблизительно на 80%. Клиническая значимость взаимодействия с неомицином не известна, но оно может являться причиной снижения эффективности регорафениба.
Комплексообразующие соединения солей желчных кислот, такие как колестирамин и холестагель, могут взаимодействовать с регорафенибом, образуя нерастворимые комплексы, которые оказывают влияние на абсорбцию (или реабсорбцию), что потенциально может привести к снижению экспозиции. Клиническое значение данных потенциальных взаимодействий неизвестно, но может привести к снижению эффективности регорафениба.
Стиварга — инструкция по применению
Синонимы, аналоги
Статьи
Регистрационный номер: ЛП-003405
Торговое наименование препарата: Стиварга®/Stivarga®
Международное непатентованное наименование: Регорафениб/ Regorafenib
Лекарственная форма: таблетки, покрытые пленочной оболочкой
Состав:
Одна таблетка, покрытая пленочной оболочкой, содержит:
Действующее вещество: регорафениб — 40,00 мг.
Вспомогательные вещества: целлюлоза микрокристаллическая — 100,00 мг, кроскармеллоза натрия — 154,00 мг, магния стеарат — 3,60 мг, повидон-25 — 160,00 мг, кремния диоксид коллоидный — 2,40 мг.
Пленочная оболочка: опадрай II™ 85G35294 розовый (железа оксид красный Е172 — 0,0648 мг, железа оксид желтый Е172 — 0,0732 мг, лецитин — 0,42 мг, макрогол/PEG 3350 — 1,482 мг, поливиниловый спирт, частично гидролизованный — 5,28 мг, тальк — 2,40 мг, титана диоксид Е171- 2,28 мг) — 12,00 мг.
Описание
Овальные таблетки, покрытые пленочной оболочкой, светло-розового цвета, на одной стороне методом выдавливания нанесено «40», на другой стороне — «BAYER».
Фармакотерапевтическая группа
Противоопухолевый препарат, ингибитор протеинкиназы.
Код АТХ: L01XE21
Фармакологические свойства
Фармакодинамика
Механизм действия
Регорафениб является ингибитором многочисленных протеинкиназ, включая киназы, участвующие в ангиогенезе опухоли (VEGFR1,-2,-3, TIE2), онкогенезе (KIT, RET, RAF-1, BRAF, BRAFv600E), а также входящие в состав микроокружения опухоли (PDGFR, FGFR). В частности, регорафениб ингибирует мутантную киназу KIT, ключевой онкогенный фактор в развитии стромальных опухолей желудочно-кишечного тракта. Благодаря этому регорафениб блокирует пролиферацию опухолевых клеток. В доклинических исследованиях было показано, что регорафениб оказывает выраженное противоопухолевое действие на широком спектре опухолевых моделей, включая колоректальный рак и гастроинтестинальные стромальные опухоли. Эффект препарата связан с его антиангиогенным и антипролиферативным воздействием. Кроме того, регорафениб показывает антиметастатическое действие in vivo. Основные метаболиты препарата в организме человека (М-2 и М-5) по своей эффективности на моделях in vitro и in vivo сопоставимы с регорафенибом.
Фармакокинетика
Всасывание
После приема таблеток регорафениба его средняя относительная биодоступность по сравнению с раствором для приема внутрь составляет 69-83 %.
Среднее значение пикового уровня регорафениба в плазме крови (Cmax) составляет около 2,5 мг/л приблизительно через 3-4 часа после однократной пероральной дозы регорафениба 160 мг (4 таблетки по 40 мг).
Наибольшая концентрация регорафениба и его основных фармакологически активных метаболитов М-2 (N-оксид) и М-5 (N-оксид и N-дезметил) достигается после приема завтрака с низким содержанием жиров по сравнению с приемом после завтрака с высоким содержанием жиров или приемом натощак. По сравнению с приемом натощак экспозиция регорафениба увеличивается на 48 % при приеме после завтрака с высоким содержанием жиров и на 36 % при приеме после завтрака с низким содержанием жиров. По сравнению с приемом натощак, экспозиция метаболитов М-2 и М-5 выше при приеме регорафениба после завтрака с низким содержанием жиров и ниже при приеме после завтрака с высоким содержанием жира.
Распределение
Кривая зависимости «концентрация-время» демонстрирует несколько пиков как для регорафениба, так и для его основных циркулирующих метаболитов, в течение 24 часов после приема дозы, что связано с печеночно-кишечной рециркуляцией препарата. Связь регорафениба с белками плазмы крови in vitro высокая и составляет 99,5 %.
Метаболизм
Метаболизм регорафениба осуществляется, главным образом, в печени путем окисления, опосредованного изоферментом CYP3A4, а также путем глюкуронирования, опосредованного UGT1A9. В плазме выявляются два основных и шесть второстепенных метаболитов регорафениба. Основные циркулирующие в плазме крови метаболиты регорафениба М-2 (N-оксид) и M-5 (N-оксид и N-дезметил) обладают фармакологической активностью и в равновесном состоянии имеют концентрации, сходные с концентрацией регорафениба.
In vitro связь М-2 и М-5 с белками крови выше, чем у регорафениба и составляет 99,8 % и 99,95 % соответственно.
Метаболиты могут быть восстановлены и гидролизированы микрофлорой желудочнокишечного тракта, при этом возможно повторное всасывание неконъюгированного препарата и его метаболитов (печеночно-кишечная рециркуляция).
Выведение
Период полувыведения регорафениба и его метаболита М-2 из плазмы составляет от 20 до 30 часов после приема внутрь. Средний период полувыведения метаболита М-5 составляет около 60 часов (40 — 100 часов).
Приблизительно 90 % дозы препарата, меченого радиоактивным изотопом, выводится в течение 12 дней после его приема, при этом 71 % выводится через кишечник (47 % в виде исходного соединения и 24 % в виде метаболитов) и около 19 % — почками в виде глюкуронидов. В равновесном состоянии выведение глюкуронидов почками уменьшается и составляет меньше 10 %. Исходное соединение, обнаруженное в каловых массах, может быть продуктом желудочно-кишечного расщепления глюкуронидов, или продуктом восстановления метаболита М-2 (N-оксида), или остатком неабсорбированного препарата.
Линейность/нелинейность
Системное воздействие регорафениба при равновесной концентрации возрастает пропорционально дозе при дозе до 60 мг и менее пропорционально при дозах более 60 мг. Накопление препарата при равновесной концентрации приблизительно в два раза превышает концентрацию в плазме, которая соответствует периоду полувыведения и частоте приема лекарственного средства.
После перорального приема регорафениба в дозе 160 мг его средняя максимальная концентрация в плазме крови (Cmax) в равновесном состоянии достигает 3,9 мг/л (8,1 мкмоль). Соотношение максимальной и минимальной концентрации регорафениба в плазме крови составляет менее 2.
Для обоих метаболитов М-2 и М-5 свойственно нелинейное накопление в плазме крови. После однократного приема регорафениба концентрации метаболитов М-2 и М-5 намного ниже, чем у исходного соединения. В равновесном состоянии концентрации М-2 и М-5 сопоставимы с концентрацией регорафениба.
Фармакокинетика у различных групп пациентов
Пациенты с нарушением функции печени
У пациентов с легкой (класс А по классификации Чайлд-Пью) или умеренной (класс В по классификации Чайлд-Пью) степенью печеночной недостаточности, фармакокинетические параметры регорафениба были такими же, как у пациентов с нормальной печеночной функцией. У пациентов с тяжелой степенью печеночной недостаточности (класс С по классификации Чайлд-Пью) фармакокинетика регорафениба не изучена. Поскольку печень имеет большое значение в выведении регорафениба, у пациентов с тяжелым нарушением функции печени возможно усиление действия препарата (см. раздел «Особые указания»).
Пациенты с нарушением функции почек
У пациентов с легкой и средней степенью почечной недостаточности экспозиция регорафениба и его метаболитов М-2 и М-5 в равновесном состоянии является такой же, как у пациентов с нормальной функцией почек. У пациентов с тяжелой степенью почечной недостаточности или терминальной степенью почечной недостаточности фармакокинетика регорафениба не изучена.
Пациенты пожилого возраста
Влияние возраста на фармакокинетику регорафениба у пациентов от 29 до 85 лет не обнаружено.
Пол
Не выявлено различий в фармакокинетике регорафениба в зависимости от пола.
Этническая принадлежность
Не выявлено различий в фармакокинетических параметрах регорафениба в зависимости от принадлежности к этнической группе.
Электрофизиология сердца/удлинение QТ
У пациентов с онкологическими заболеваниями не было выявлено удлинение интервала QТ в равновесном состоянии при приеме регорафениба в дозе 160 мг.
Показания к применению
Противопоказания
С осторожностью
Необходимо соблюдать дополнительную осторожность при назначении препарата в следующих ситуациях:
Применение при беременности и в период грудного вскармливания
Фертильность
Не проводилось специальных исследований применения регорафениба для оценки его влияния на фертильность человека.
В исследованиях на животных наблюдалось снижение мужской и женской фертильности. Женщин репродуктивного возраста необходимо проинформировать об опасности препарата Стиварга® для плода. Во время лечения и в течение 8 недель после терапии препаратом Стиварга® женщины и мужчины репродуктивного возраста должны применять надежные методы контрацепции.
Беременность
Данные о применении препарата Стиварга® у беременных женщин отсутствуют.
Учитывая механизм действия регорафениба, возможно негативное влияние препарата Стиварга® на плод. Исследования на животных продемонстрировали репродуктивную токсичность регорафениба.
Период грудного вскармливания
Не установлено, выделяются ли регорафениб и его метаболиты с женским молоком.
Исследования на животных показали, что регорафениб и его метаболиты выделяются с грудным молоком.
Поскольку нельзя исключить возможность негативного влияния регорафениба на рост и развитие детей раннего возраста, следует прекратить грудное вскармливание в период лечения препаратом Стиварга®.
Способ применения и дозы
Для приема внутрь.
Препарат Стиварга® должен назначаться только врачом, имеющим опыт противоопухолевой терапии.
Рекомендуемая суточная доза препарата Стиварга® составляет 160 мг (4 таблетки по 40 мг). Препарат назначается один раз в сутки в течение 3 недель. В последующую неделю (4-я неделя от начала лечения) следует перерыв в приеме препарата. Период продолжительностью 4 недели от начала приема является одним курсом лечения препаратом Стиварга®.
Таблетки принимают каждый день (один раз в сутки) в одно и то же время после приема пищи, содержащей низкое (< 30 %) количество жира (см. раздел «Фармакокинетика»). Таблетки следует проглатывать целиком, запивая водой.
Если очередной прием препарата пропущен, пациент должен принять таблетку в тот же день, как только об этом вспомнит. Не следует принимать двойную дозу препарата в течение одного дня с целью компенсировать пропущенный прием.
В случае рвоты после приема препарата Стиварга® дополнительную таблетку принимать не следует.
Лечение продолжают до тех пор, пока сохраняется клиническая эффективность препарата или до появления его неприемлемого токсического действия (см. раздел «Особые указания»).
Коррекция дозы
Индивидуальная переносимость и безопасность лечения может потребовать временного прекращения терапии и/или уменьшения дозы препарата Стиварга®.
Коррекция дозы на каждом этапе снижения дозы составляет 40 мг (1 таблетка). Наименьшая рекомендуемая доза препарата Стиварга® составляет 80 мг в сутки. Максимальная суточная доза 160 мг.
Таблица 1.
Рекомендации по коррекции дозы препарата Стиварга® при развитии ладонно-подошвенной эритродизестезии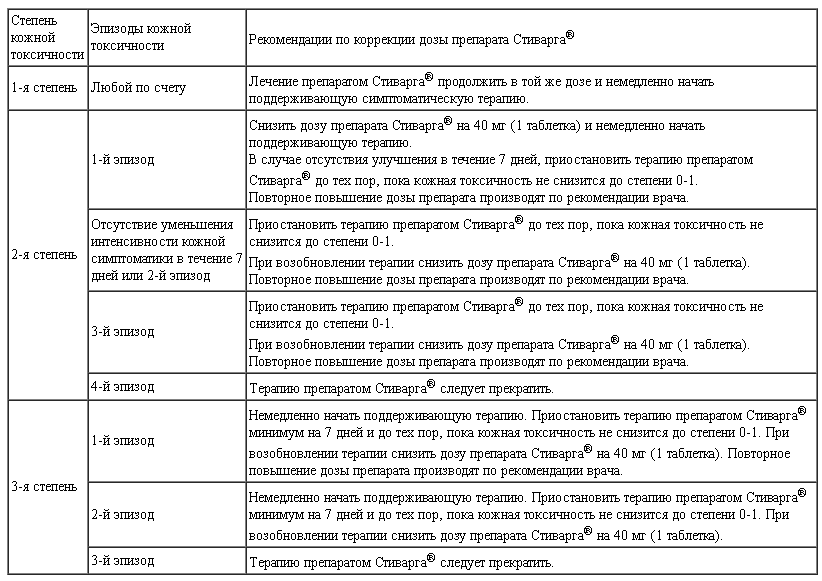
Таблица 2.
Рекомендации по коррекции дозы препарата Стиварга® при ухудшении биохимических показателей функции печени (см. раздел «Особые указания»)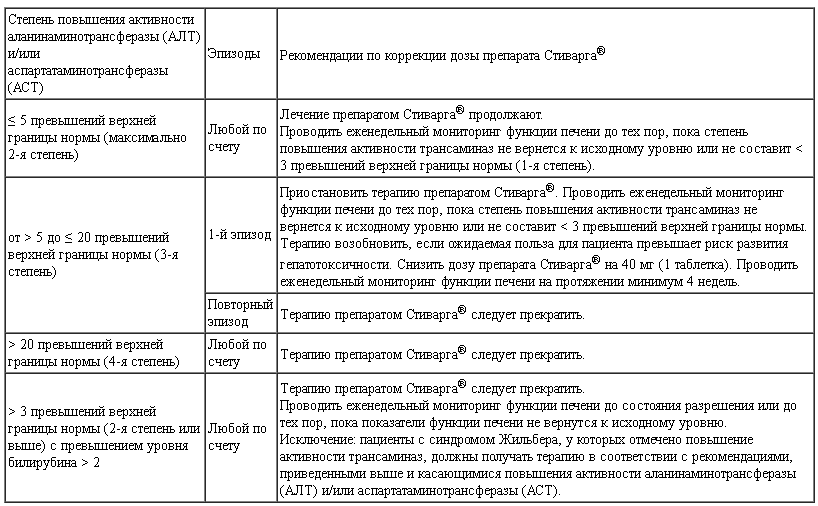
Особые группы пациентов
Пациенты с нарушением функции печени
Печень имеет большое значение в выведении регорафениба. Не было выявлено клинически значимых различий в воздействии препарата Стиварга® у пациентов со средним (класс А по классификации Чайлд-Пью) и умеренным (класс В по классификации Чайлд-Пью) нарушением функции печени в сравнении с пациентами с нормальной функцией печени. Пациентам со средним и умеренным нарушением функции печени коррекция дозы не требуется. Рекомендуется проводить мониторинг функции печени на фоне терапии препаратом Стиварга® у данной категории пациентов (см. разделы «Фармакокинетика», «Особые указания»). Не рекомендуется лечение препаратом Стиварга® пациентов с тяжелым нарушением функции печени (класс С по классификации Чайлд-Пью), поскольку применение препарата у данной категории пациентов не изучено.
Пациенты с нарушением функции почек
В клинических исследованиях не отмечалось значимых различий безопасности и эффективности препарата Стиварга® у пациентов с легкой степенью почечной недостаточности (рСКФ 60-89 мл/мин/1,73 м2) в сравнении с пациентами с нормальной функцией почек. Ограниченные данные по фармакокинетике не выявили различий в экспозиции у пациентов со средней степенью почечной недостаточности (рСКФ 30-59 мл/мин/1,73 м2). Пациентам с легкой и средней степенью почечной недостаточности снижение дозы препарата не требуется (см. раздел «Фармакокинетика»). Применение препарата Стиварга® у пациентов с тяжелой степенью почечной недостаточности (рСКФ < 30 мл/мин/1,73 м2) не изучено.
Дети
Безопасность и эффективность назначения препарата Стиварга® у детей и подростков до 18 лет не установлена.
Пациенты пожилого возраста
В клинических исследованиях не отмечалось значимых различий безопасности и эффективности препарата Стиварга® у пожилых (65 лет и старше) пациентов в сравнении с более молодыми пациентами. Пациентам пожилого возраста коррекция дозы не требуется (см. раздел «Фармакокинетика»).
Пол
В клинических исследованиях не отмечалось значимых различий безопасности и эффективности препарата Стиварга® между пациентами мужского и женского пола. Коррекция дозы препарата в зависимости от пола пациента не требуется (см. раздел «Фармакокинетика»).
Этническая принадлежность
В клинических исследованиях не отмечалось значимых различий безопасности и эффективности препарата Стиварга® у пациентов разных этнических групп. Корректировка дозы препарата в зависимости от этнической принадлежности пациента не требуется (см. раздел «Фармакокинетика»).
Побочное действие
Общий профиль безопасности препарата Стиварга® оценивается на данных клинических исследований с участием более 1200 пациентов, получавших терапию в плацебо-контролируемых клинических исследованиях III фазы. Данная группа включала 500 пациентов с метастатическим колоректальным раком и 132 пациента с гастроинтестинальными стромальными опухолями.
Данный профиль безопасности регорафениба был подтвержден и в клиническом исследовании III В фазы с участием 2872 пациентов с метастатическим колоректальным раком при прогрессировании на стандартной терапии.
В клинических исследованиях наиболее частыми нежелательными реакциями (≥ 30% пациентов) были астения/усталость, ладонно-подошвенная эритродизестезия, диарея, снижение аппетита и потребления пищи, повышение артериального давления, дисфония, инфекции.
Наиболее серьезными нежелательными реакциями при приеме препарата Стиварга® были поражения печени, кровотечения, прободение желудочно-кишечного тракта.
Перечисленные ниже нежелательные явления, отмеченные при применении препарата Стиварга® в ходе клинических исследований, распределены по частоте возникновения в соответствии со следующей градацией: очень часто (≥1/10), часто (от ≥1/100 до <1/10), нечасто (от ≥ 1/1000 до <1/100), редко (от ≥ 1/10000 до <1/1000). Для классификации и описания конкретной реакции, ее синонимов и связанных с ней состояний, используется наиболее подходящий термин из Медицинского словаря для регуляторной деятельности (MedDRA).
В каждой частотной группе нежелательные явления представлены в порядке уменьшения их значимости.
Нарушения со стороны крови и лимфатической системы
Очень часто: тромбоцитопения, анемия.
Часто: лейкопения.
Нарушения со стороны сердца и сосудов
Очень часто: кровотечения*, повышение артериального давления.
Нечасто: инфаркт миокарда, ишемия миокарда, гипертонический криз.
Нарушения со стороны дыхательной системы, органов грудной клетки и средостения
Очень часто: дисфония.
Нарушения со стороны кожи и подкожных тканей
Очень часто: ладонно-подошвенная эритродизестезия, кожная сыпь, алопеция.
Часто: сухость кожи, эксфолиативный дерматит.
Нечасто: поражение ногтей, мультиформная эритема.
Редко: синдром Стивенса-Джонсона, токсический эпидермальный некролиз.
Нарушения со стороны желудочно-кишечного тракта
Очень часто: диарея, стоматит, рвота, тошнота.
Часто: нарушение вкуса, сухость слизистой оболочки полости рта, гастроэзофагеальный рефлюкс, гастроэнтерит.
Нечасто: прободение желудочно-кишечного тракта*, свищ желудочно-кишечного тракта.
Нарушения со стороны печени и желчевыводящих путей
Очень часто: гипербилирубинемия.
Часто: повышение активности трансаминаз.
Нечасто: тяжелое нарушение функции печени*#.
Нарушения со стороны нервной системы
Очень часто: головная боль.
Часто: тремор.
Редко: синдром задней обратимой энцефалопатии.
Нарушения со стороны скелетно-мышечной и соединительной ткани
Часто: мышечно-скелетная ригидность.
Нарушения со стороны почек и мочевыводящих путей
Часто: протеинурия.
Нарушения со стороны эндокринной системы
Часто: гипотиреоз.
Нарушения со стороны обмена веществ и питания
Очень часто: снижение аппетита и потребления пищи.
Часто: гипокалиемия, гипофосфатемия, гипокальциемия, гипонатриемия, гипомагниемия, гиперурикемия.
Лабораторные и инструментальные данные
Очень часто: снижение массы тела.
Часто: увеличение активности амилазы и липазы, отклонение от нормального значения международного нормализованного отношения (МНО).
Доброкачественные, злокачественные и неуточненные новообразования (включая кисты и полипы)
Редко: кератоакантома/ плоскоклеточный рак кожи.
Инфекционные и паразитарные заболевания
Очень часто: инфекции.
Общие расстройства и нарушения в месте введения
Очень часто: астения/общая слабость, боль различной локализации, повышение температуры тела, воспаление слизистых оболочек.
* сообщалось о летальном исходе в результате неблагоприятной реакции;
# в соответствии с критериями Международной рабочей группы экспертов по лекарственному поражению печени.
Поражения печени
В большинстве случаев тяжелого поражения печени дисфункция печени начиналась в течение первых двух месяцев терапии и характеризовалась повреждением гепатоцитов с повышением активности трансаминаз > 20 х ВГН (верхняя граница нормы), сопровождаемым увеличением концентрации билирубина. В клинических исследованиях тяжелые поражения печени со смертельным исходом чаще наблюдались у пациентов японской национальности, получавших препарат Стиварга® (около 1,5 %), в сравнении с пациентами не японской национальности (<0,1 %).
Кровотечения
В двух плацебо-контролируемых исследованиях III фазы общая частота кровотечений у пациентов, получавших препарат Стиварга®, составила 19,3 %. Большинство случаев кровотечения имели легкую или умеренную степень тяжести (1-я и 2-я степень: 16,9 %). Наиболее часто отмечалось носовое кровотечение (7,6 %). Летальные исходы у пациентов, получавших препарат Стиварга®, отмечались редко (0,6 %) и чаще были связаны с поражением дыхательной, пищеварительной и мочеполовой систем.
Инфекции
В двух плацебо-контролируемых исследованиях III фазы инфекционные заболевания чаще отмечались у пациентов, получавших лечение препаратом Стиварга®, по сравнению с пациентами, получавшими плацебо (все степени: 31,0 % по сравнению с 14,4 %). Чаще инфекции у пациентов, получавших препарат Стиварга®, имели легкую или умеренную степень выраженности (1-я и 2-я степень: 22,9 %) и включали инфекции мочевыводящих путей (6,8 %), назофарингит (4,2 %), а также кандидоз кожи и слизистых и системный микоз (2,4 %). Не наблюдалось различий по частоте летальных исходов в связи с развитием инфекции у пациентов, получавших препарат Стиварга® (0,6 %) и у пациентов, получавших плацебо (0,6 %).
Ладонно-подошвенная эритродизестезия
В плацебо-контролируемом исследовании III фазы общая частота возникновения ладонноподошвенной эритродизестезии у пациентов с метастатическим колоректальным раком, принимавших препарат Стиварга®, составила 45,2 % и у пациентов, получающих плацебо — 7,1 %. В плацебо-контролируемом исследовании III фазы у пациентов с гастроинтестинальными стромальными опухолями общая частота возникновения ладонно-подошвенной эритродизестезии составила 66,7 % у пациентов, получавших терапию препаратом Стиварга®, и 15,2 % у пациентов, получавших плацебо. В обоих исследованиях большинство случаев ладонно-подошвенной эритродизестезии у пациентов, получавших препарат Стиварга®, отмечалось во время первого цикла лечения и имело легкую или среднюю степень тяжести (1-я и 2-я степени: 28,6 % у пациентов с метастатическим колоректальным раком и 44,7 % у пациентов с гастроинтестинальными стромальными опухолями). Частота возникновения ладонно-подошвенной эритродизестезии 3-й степени составила 16,6 % у пациентов с метастатическим колоректальным раком и 22,0 % у пациентов с гастроинтестинальными стромальными опухолями.
Повышение артериального давления
В плацебо-контролируемом исследовании III фазы у пациентов с метастатическим колоректальным раком общая частота случаев повышения артериального давления составила 30,4 % у пациентов, принимавших препарат Стиварга®, и 7,9 % у пациентов, принимавших плацебо. В плацебо-контролируемом исследовании III фазы у пациентов с гастроинтестинальными стромальными опухолями общая частота случаев повышения артериального давления составила 59,1 % у пациентов, получавших препарат Стиварга®, и 27,3 % у пациентов, получавших плацебо. В обоих исследованиях большинство случаев повышения артериального давления у пациентов, получавших препарат Стиварга®, было зарегистрировано в течение первого цикла лечения. При этом случаи повышения артериального давления имели легкую и умеренную степень тяжести (1-я и 2-я степень: 22,8 % у пациентов с метастатическим колоректальным раком и 31,1 % у пациентов с гастроинтестинальными стромальными опухолями). Частота случаев повышения артериального давления 2-й степени составила 7,6 % (у пациентов с колоректальным раком) и 27,3 % (у пациентов с гастроинтестинальными стромальными опухолями). Был зарегистрирован один случай повышения артериального давления 4-й степени у пациента с гастроинтестинальной стромальной опухолью.
Протеинурия
В плацебо-контролируемом исследовании III фазы среди пациентов с метастатическим колоректальным раком частота случаев протеинурии, связанной с лечением, составила 7.4 % у пациентов, получавших терапию препаратом Стиварга®, в сравнении с 2,4 % у пациентов, принимавших плацебо. После развития протеинурии у 40,5 % пациентов из группы препарата Стиварга® и 66,7 % пациентов из группы плацебо состояние не вернулось к исходным значением. В плацебо-контролируемом исследовании III фазы среди пациентов с гастроинтестинальными стромальными опухолями общая частота случаев протеинурии составила 6,8 % у пациентов, получающих терапию препаратом Стиварга®, в сравнении с 1,5% у пациентов, принимавших плацебо.
Нарушения со стороны сердца и сосудов
Во всех проводимых клинических исследованиях нежелательные явления в виде нарушений со стороны сердца (все степени) чаще регистрировались (20,5 % относительно 10.4 %) у пациентов, получавших терапию препаратом Стиварга®, в возрасте 75 лет или старше (N=78), чем у пациентов, получавших терапию препаратом Стиварга®, в возрасте до 75 лет (N=995).
Лабораторные и инструментальные данные
В двух плацебо-контролируемых исследованиях III фазы у 26,1 % пациентов, получавших терапию препаратом Стиварга®, и у 15,1 % пациентов, получавших плацебо, концентрация тиреотропного гормона (ТТГ) была выше верхней границы нормы. Значения ТТГ в 4 раза превышающие верхнюю границу нормы были зарегистрированы у 6,9 % пациентов, получавших терапию препаратом Стиварга®, и у 0,7 % пациентов, принимавших плацебо. Концентрация свободного трийодтиронина (Т3 свободный) меньше нижней границы нормы отмечалась у 25,6 % пациентов, получающих терапию Стиварга®, и у 20,9 % пациентов, получающих плацебо. Концентрация свободного тироксина (Т4 свободный) меньше нижней границы нормы отмечалась у 8,0 % пациентов в группе терапии препаратом Стиварга® и у 6,6 % пациентов из группы плацебо. В целом, приблизительно у 7 % пациентов, получающих терапию препаратом Стиварга®, развился гипотиреоз, требующий применения заместительной гормонотерапии.
Передозировка
Симптомы
В клинических исследованиях максимальная суточная доза препарата Стиварга® составляла 220 мг. Наиболее частыми нежелательными реакциями при данной дозе препарата были нежелательные реакции со стороны кожи, дисфония, диарея, воспаление слизистых оболочек, сухость слизистой оболочки полости рта, снижение аппетита, повышение артериального давления, общая слабость.
Лечение
Специфический антидот не известен.
В случае передозировки прием препарата Стиварга® следует прекратить и применять стандартную симптоматическую терапию. Пациент должен находиться под наблюдением врача до стабилизации состояния.
Взаимодействие с другими лекарственными препаратами и другие формы взаимодействия
Фармакокинетические взаимодействия
Индукторы CYP3A4/ингибиторы CYP3A4 и UGT1A9
In vitro было показано, что регорафениб метаболизируется цитохромом CYP3A4 и уридинфосфатглюкуронилтрансферазой UGT1A9.
Применение кетоконазола (400 мг в течение 18 дней), сильного ингибитора изофермента CYP3A4, в сочетании с однократным приемом регорафениба (160 мг на пятый день) приводило к увеличению среднего воздействия (AUC) регорафениба приблизительно на 33% и снижению среднего воздействия его активных метаболитов, М-2 (N-оксид) и M-5 (N-оксид и N-дезметил) приблизительно на 90 %. Не рекомендуется применять препарат Стиварга® совместно с сильными ингибиторами CYP3A4 (например, кларитромицин, грейпфрутовый сок, итраконазол, кетоконазол, позаконазол, телитромицин и вориконазол), поскольку их влияние на воздействие регорафениба в стабильном состоянии и его метаболитов (М-2 и М-5) не изучалось.
Не рекомендуется применять сильные ингибиторы UGT1A9 (например, мефенаминовая кислота, дифлунизал и нифлумовая кислота) во время терапии регорафенибом, поскольку их влияние на экспозицию регорафениба и его метаболитов в равновесном состоянии не изучалось.
Применение рифампина (600 мг в течение 9 дней), сильного индуктора CYP3A4, в сочетании с однократным приемом регорафениба (160 мг на седьмой день) приводило к снижению среднего воздействия (AUC) регорафениба приблизительно на 50 %, увеличению среднего воздействия активного метаболита М-5 в 3-4 раза, при этом не отмечалось изменения экспозиции активного метаболита М-2. Другие сильные ингибиторы CYP3A4 (например, фенитоин, карбамазепин, фенобарбитал) могут увеличивать метаболизм регорафениба. Не рекомендуется применять препарат Стиварга® совместно с сильными индукторами CYP3A4 или подбирать лекарственные средства, которые не влияют на CYP3A4 или индуцируют его в минимальной степени.
Субстраты UGT1A1 и UGT1A9
In vitro было показано, что регорафениб, также как и его активный метаболит М-2, подавляет глюкуронизацию под действием UGT1A1 и UGT1A9, в то время как метаболит M-5 подавляет UGT1A1 только в концентрациях, которые достигаются в равновесном состоянии in vivo.
Применение регорафениба с последующим 5-дневным перерывом перед назначением иринотекана приводило к увеличению среднего воздействия (AUC) SN-38, субстрата UGT1A1 и активного метаболита иринотекана приблизительно на 44%. Также отмечалось увеличение среднего воздействия (AUC) иринотекана приблизительно на 28%. Эти данные показывают, что комбинированное применение регорафениба может увеличивать системную экспозицию субстратов UGT1A1 и UGT1A9.
Субстраты белка резистентности рака молочной железы (BCRP) и P-гликопротеина
Применение регорафениба (160 мг в течение 14 дней) перед однократным приемом розувастатина (5 мг), который является субстратом BCRP, приводило к увеличению среднего воздействия (AUC) розувастатина в 3,8 раз и к увеличению его средней максимальной концентрации в плазме крови (Cmax) в 4,6 раза. Комбинированное применение с регорафенибом может повышать концентрацию в плазме других субстратов BCRP (например, метотрексат, флувастатин, аторвастатин). Рекомендуется контролировать состояние пациентов с целью выявления признаков и симптомов увеличения экспозиции к субстратам BCRP.
В клинических исследованиях было показано, что регорафениб не оказывает влияния на фармакокинетические параметры дигоксина, поэтому его можно применять совместно с субтратами Р-гликопротеина, такими как дигоксин. При этом не отмечается клинически значимого взаимодействия между лекарственными препаратами.
Ингибиторы Р-гликопротеина и BCRP/стимуляторы Р-гликопротеина и BCRP
Исследования in vitro показывают, что метаболиты регорафениба М-2 и М-5 являются субстратами Р-гликопротеина и BCRP. Ингибиторы и стимуляторы BCRP и Р-гликопротеина могут препятствовать экспозиции М-2 и М-5. Клиническая значимость данных результатов исследований неизвестна.
Селективные субстраты изоформы CYP
In vitro было показано, что регорафениб является конкурентоспособным ингибитором цитохромов CYP2C8, CYP2C9, CYP2B6 в концентрациях, которые достигаются in vivo в стабильном состоянии (максимальная концентрация в плазме 8,1 мкмоль). In vitro ингибирующее действие в отношении CYP3A4 и CYP2C19 менее выражено.
Было проведено исследование с целью оценки влияния приема регорафениба в дозе 160 мг в течение 14 дней на фармакокинетику маркерных субстратов CYP2C8 (розиглитазон), CYP2C9 (варфарин), CYP2C19 (омепразол) и CYP3A4 (мидазолам).
Фармакокинетические данные показывают, что регорафениб можно применять совместно с субстратами CYP2C8, CYP2C9, CYP3A4 и CYP2C19. При этом не отмечается клинически значимого взаимодействия между лекарственными препаратами.
Антибиотики
Профиль концентрация-время показывает, что регорафениб и его метаболиты могут подвергаться печеночно-кишечной циркуляции (см. раздел «Фармакокинетика»). Комбинированное применение регорафениба с неомицином (слабо абсорбируемый противомикробный препарат, применяемый с целью эрадикации флоры желудочнокишечного тракта) не оказывает влияние на экспозицию регорафениба, но может нарушить его печеночно-кишечную циркуляцию. Тем не менее при сравнении фармакологической активности регорафениба in vivo и in vitro было показано уменьшение экспозиции его активных метаболитов M-2 и M-5 приблизительно на 80 %. Клиническая значимость взаимодействия с неомицином не известна, но оно может являться причиной снижения эффективности регорафениба. Фармакокинетическое взаимодействие регорафениба с другими антибиотиками не изучалось.
Комплексообразующие соединения солей желчных кислот
Существует вероятность, что регорафениб и его метаболиты М-2 и М-5 могут подвергаться печеночно-кишечной циркуляции (см. раздел «Фармакокинетика). Комплексообразующие соединения солей желчных кислот, такие как холестирамин и холестагель могут взаимодействовать с регорафенибом, образуя нерастворимые комплексы, которые оказывают влияние на абсорбцию (или реабсорбцию), что потенциально может привести к снижению экспозиции. Клиническое значение данных потенциальных взаимодействий неизвестно, но может привести к снижению эффективности регорафениба.
Особые указания
Действие на печень
У пациентов, получавших лечение препаратом Стиварга®, часто регистрировались отклонения значений биохимических показателей функции печени (аланинаминотрансфераза (АЛТ), аспартатаминотрансфераза (АСТ) и билирубин). У небольшой части пациентов отмечались тяжелые нарушения показателей функции печени (3-4 степени тяжести) и клинически выраженные нарушения функции печени (в том числе с летальным исходом) (см. раздел «Побочное действие»).
До начала лечения препаратом Стиварга® рекомендуется определить показатели функции печени (АСТ, АЛТ, билирубин). На протяжении первых двух месяцев терапии следует проводить контроль функции печени по меньшей мере каждые 2 недели, затем не реже 1 раза в месяц, а также согласно клиническим показателям.
Поскольку регорафениб является ингибитором уридиндифосфатглюкуронилтрансферазы (UGT1A1), у пациентов с синдромом Жильбера возможно появление слабовыраженной непрямой (неконъюгированной) гипербилирубинемии (см. раздел «Взаимодействие с другими лекарственными препаратами и другие формы взаимодействия»).
Если у больных, получающих лечение препаратом Стиварга®, отмечается ухудшение показателей функции печени, связанное с терапией (в отсутствие явной альтернативной причины, например, механической желтухи или прогрессирования основного заболевания), врачу следует изменить дозу препарата и наблюдать за состоянием пациента (см. раздел «Способ применения и дозы», таблица 1).
Печень имеет большое значение в выведении регорафениба. При применении препарата Стиварга® у пациентов с легким и умеренным нарушением функции печени следует проводить тщательный контроль за состоянием пациента (см. раздел «Способ применения и дозы»). Не рекомендуется применение препарата Стиварга® у пациентов с тяжелым нарушением функции печени (класс С по классификации Чайлд-Пью), поскольку препарат Стиварга® не изучен у данной категории пациентов (возможно увеличение экспозиции препарата).
Кровотечения
У больных, получавших препарат Стиварга®, было зарегистрировано повышение частоты эпизодов кровотечений, в некоторых случаях с летальным исходом (см. раздел «Побочное действие”). При наличии факторов риска кровотечения, а также при совместном назначении с антикоагулянтами (например, варфарин, фенпрокумон) или другими лекарственными препаратами, повышающими риск кровотечения, следует контролировать показатели коагулограммы и общего анализа крови. При появлении тяжелого кровотечения, требующего экстренного медицинского вмешательства, следует рассмотреть вопрос о прекращении лечения препаратом Стиварга®.
Ишемия миокарда и инфаркт миокарда
При приеме препарата Стиварга® отмечалось увеличение частоты случаев ишемии миокарда и инфаркта миокарда (см. раздел «Побочное действие”).
Пациенты с нестабильной стенокардией или появлением стенокардии (в течение 3 месяцев до начала терапии препаратом Стиварга®), недавним инфарктом миокарда (в период 6 месяцев до начала терапии препаратом Стиварга®) и пациенты с сердечной недостаточностью 2 класса и выше по классификации Нью-Йоркской кардиологической ассоциации были исключены из клинических исследований.
У пациентов с ишемической болезнью сердца необходимо отслеживать клинические признаки и симптомы ишемии миокарда. При возникновении ишемии и/или инфаркта миокарда следует прекратить терапию препаратом Стиварга® до нормализации состояния. При принятии решения о возобновлении терапии врач должен оценивать соотношение пользы от приема препарата с потенциальным риском у каждого отдельного пациента. Если клинические проявления ишемии сохраняются, возобновлять терапию не следует.
Синдром обратимой задней энцефалопатии.
У пациентов, получавших препарат Стиварга®, были зарегистрированы случаи развития обратимой задней энцефалопатии (см. раздел «Побочное действие”). Обратимая задняя энцефалопатия проявлялась в виде судорог, головной боли, изменения сознания, нарушения зрения или корковой слепоты, иногда в сочетании с артериальной гипертензией. Для подтверждения диагноза пациентам следует провести томографию головного мозга. В случае развития обратимой задней энцефалопатии следует прекратить лечение препаратом Стиварга®, проводить контроль артериального давления и поддерживающую терапию.
Прободение и свищ желудочно-кишечного тракта
У пациентов, получавших препарат Стиварга®, были зарегистрированы случаи прободения желудочно-кишечного тракта и образования свища желудочно-кишечного тракта (см. раздел «Побочное действие»). Эти события были связаны с опухолями в брюшной полости. В случае прободения желудочно-кишечного тракта или образования свища терапию препаратом Стиварга® следует прекратить.
Повышение артериального давления
На фоне терапии препаратом Стиварга® было зарегистрировано увеличение частоты повышения артериального давления (см. раздел «Побочное действие»). Перед началом и во время лечения препаратом Стиварга® следует регулярно контролировать артериальное давление и корректировать его повышение в соответствии с принятыми стандартами лечения. В случаях развития тяжелой или стойкой артериальной гипертензии, устойчивой к проводимой адекватной антигипертензивной терапии, врач должен временно прервать терапию и/или снизить дозу препарата (см. раздел «Способ применения и дозы”). В случае развития гипертонического криза лечение препаратом Стиварга® следует отменить.
Нарушения заживления ран
В случае проведения обширных хирургических вмешательств рекомендуется временное прекращение терапии препаратом Стиварга®, поскольку лекарственные препараты, обладающие антиангиогенными свойствами, могут подавлять или ухудшать заживление ран. Решение о возобновлении терапии после хирургических вмешательств должно основываться на клинической оценке адекватности заживления раны.
Кожная токсичность
Наиболее частыми нежелательными реакциями при приеме препарата Стиварга® были ладонно-подошвенная эритродизестезия и сыпь (см. раздел «Побочное действие»). С целью профилактики развития ладонно-подошвенной эритродизестезии следует контролировать образование мозолей и использовать специальные вкладыши для обуви и перчатки для предотвращения давления на подошвы и ладони. Для лечения ладонноподошвенной эритродизестезии можно использовать кератолитические кремы (например, кремы на основе мочевины, салициловой кислоты или альфа-гидроксильной кислоты, которые следует наносить только на пораженные участки кожи) и увлажняющие кремы в обильном количестве для облегчения симптомов. При необходимости временно прекращают лечение и/или снижают дозу препарата Стиварга® или, в тяжелых или повторяющихся случаях кожных реакций, терапию препаратом Стиварга® прекращают (см. раздел «Способ применения и дозы»).
Отклонения значений лабораторных показателей
При применении препарата Стиварга® было зарегистрировано повышение частоты электролитных нарушений (включая гипофосфатемию, гипокальциемию, гипонатриемию и гипокалиемию) и нарушений метаболизма (включая увеличение концентрации тиреотропного гормона, увеличение активности амилазы). Отклонения от нормы обычно носили легкий или умеренный характер и не сопровождались клиническими проявлениями. В случае возникновения электролитных нарушений или нарушений метаболизма коррекция дозы или прекращение терапии не требуется. Во время терапии препаратом Стиварга® рекомендуется контролировать биохимические и метаболические показатели. При необходимости назначают заместительную терапию в соответствии с принятыми стандартами лечения. В случаях устойчивых или рецидивирующих нарушений следует рассмотреть возможность временного прекращения лечения или снижения дозы, или полного прекращения терапии препаратом Стиварга® (см. раздел «Способ применения и дозы»).
Информация о некоторых ингредиентах
Суточная доза регорафениба 160 г содержит 2,427 ммоль (или 55,8 мг) натрия. На это следует обратить внимание пациентов, соблюдающих диету, контролирующую потребление натрия. Суточная доза регорафениба 160 г содержит 1,68 мг лецитина (получен из сои).
Системная токсичность
После повторного введения дозы у мышей, крыс и собак наблюдались нежелательные реакции со стороны ряда органов, прежде всего почек, печени, пищеварительного тракта, щитовидной железы, лимфатической/кроветворной системы, эндокринной системы, репродуктивной системы и кожи. В 26-недельном исследовании токсичности при введении повторных доз у крыс наблюдалось небольшое увеличение частоты случаев утолщения атриовентрикулярных клапанов сердца. Причиной данного явления может служить ускорение возрастного физиологического процесса. Данные реакции наблюдались при системных экспозициях, находящихся в диапазоне или ниже диапазона предполагаемой экспозиции у человека (основано на сравнении AUC).
Изменения зубов и костей, а также нежелательные явления в репродуктивной системе были наиболее выражены у молодых и растущих животных, а также у молодых крыс, что указывает на потенциальный риск для детей и подростков.
Тератогенность и эмбриотоксичность
Специальные исследования влияния регорафениба на фертильность не проводились. Следует учитывать способность регорафениба оказывать неблагоприятное воздействие на репродуктивную систему мужчин и женщин. В исследовании на крысах и собаках после многократного применения регорафениба при экспозициях ниже предполагаемой экспозиции у человека (при сравнении AUС) наблюдались морфологические изменения в яичках, яичниках и матке. Наблюдаемые изменения были обратимы только частично.
В исследовании на кроликах при экспозиции ниже предполагаемой экспозиции у человека наблюдалась эмбриотоксичность (при сравнении AUС). В основном были обнаружены нарушения формирования мочевыделительной системы, сердца и крупных сосудов, костей.
Влияние на способность управлять транспортными средствами, механизмами
Специальные исследования не проводились, однако при возникновении нежелательных явлений, которые могут влиять на указанные способности, рекомендуется избегать управления транспортными средствами и занятий другими потенциально опасными видами деятельности, требующими повышенной концентрации внимания и быстроты психомоторных реакций (до исчезновения данных симптомов).
Форма выпуска
Таблетки, покрытые пленочной оболочкой, 40 мг.
По 28 таблеток с влагопоглотителем в непрозрачном флаконе белого цвета из полиэтилена высокой плотности с завинчивающейся крышкой с герметизирующей вставкой и устройством против вскрытия флакона детьми. 1 или 3 флакона с инструкцией по применению в картонной пачке.
Условия хранения
Хранить при температуре не выше 30 °С.
Хранить в оригинальной упаковке.
Хранить в недоступном для детей месте.
Срок годности
3 года.
Не применять после истечения срока годности, указанного на упаковке.
Условия отпуска
Отпускается по рецепту.
Производитель
Байер Фарма АГ, Кайзер-Вильгельм-Аллее, 51368 Леверкузен, Германия
Bayer Pharma AG, Kaiser-Wilhelm-Allee, 51368 Leverkusen, Germany
Наименование и адрес юридического лица, на имя которого выдано регистрационное удостоверение
Байер Фарма АГ, Мюллерштрассе 178, 13353 Берлин, Германия
Bayer Pharma AG, Mullerstrasse 178, 13353 Berlin, Germany
За дополнительной информацией и с претензиями обращаться по адресу:
107113 Москва, 3-я Рыбинская ул., д.18, стр.2
Комментарии
(видны только специалистам, верифицированным редакцией МЕДИ РУ)
Противоопухолевые средства. Ингибиторы протеинкиназы. Код АТХ L01XE21.
Фармакологические свойства
Фармакодинамика
Механизм действия и фармакодинамические эффекты
Регорафениб эффективно блокирует разнообразные протеинкиназы, включая киназы, участвующие в ангиогенезе (VEGFR1, -2, -3, TIE2), онкогенезе (KIT, RET, RAF-1, BRAF, BRAFV600E), образовании метастаз (VEGFR3, PDGFR, FGFR) и активности иммунитета против опухолей (CSF1R). В частности, регорафениб ингибирует мутантную киназу KIT, ключевой онкогенный фактор в развитии гастроинтестинальных стромальных опухолей. Благодаря этому регорафениб блокирует пролиферацию опухолевых клеток. В доклинических исследованиях регорафениб продемонстрировал мощную противоопухолевую активность на широком спектре опухолевых моделей, включая модели колоректальных, гастроинтестинальных стромальных и гепатоцеллюлярных опухолей. Эффект лекарственного средства связан с его антиангиогенным и антипролиферативным воздействием. Кроме того, регорафениб уменьшал уровни макрофагов, ассоциированных с опухолью и показал антиметастатическое действие in vivo. Основные метаболиты регорафениба в организме человека (М-2 и М-5) на моделях in vitro и in vivo показывают действие, аналогичное регорафенибу.
Клиническая эффективность и безопасность
Метастатический колоректальный рак
Клиническая эффективность и безопасность лекарственного средства Стиварга® была оценена в международном многоцентровом рандомизированном двойном слепом плацебо — контролируемом исследовании III фазы (CORRECT) у пациентов с метастатическим колоректальным раком, у которых наблюдалось прогрессирование заболевания после стандартной терапии.
Первичной конечной точкой оценки эффективности являлась общая выживаемость (ОВ). Вторичными критериями являлись выживаемость без прогрессирования (ВБП), уровень объективного ответа опухоли и частота контроля заболевания.
Всего было рандомизировано 760 пациентов в соотношении 2:1. Пациенты получали либо регорафениб 160 мг перорально (4 таблетки Стиварга®, содержащие 40 мг регорафениба каждая) один раз в день (N=505) плюс оптимальная поддерживающая терапия (ОПТ) (N=255), либо плацебо плюс ОПТ, в течение 3-х недель и перерывом терапии в одну неделю. Средняя суточная доза регорафениба составила 147 мг.
Лечение продолжалось до прогрессирования заболевания или возникновения клинически неприемлемых признаков токсичности. Предварительно запланированный промежуточный анализ эффективности осуществлялся после регистрации 432 летальных случаев. Исследование было выведено из слепого метода после того, как результаты данного запланированного промежуточного анализа ОВ вышли за пределы предварительно определенных границ эффективности.
Среди 760 пациентов, средний возраст которых составлял 61 год, 61% составляли мужчины, 78% пациентов принадлежали к европеоидной расе, общее состояние всех пациентов в начале исследования оценивали как 0 или 1 по шкале общего состояния (ОС) ECOG. Во время лечения лекарственным средством Стиварга® показатель общего состояния ≥ 2 был отмечен у 11,4% пациентов. Средняя продолжительность лечения и суточная доза, а также частота коррекции дозы и снижения дозы были такими же, как у пациентов ОС ≥ 2 получавших плацебо 18,6%. Большинство пациентов с ОС ≥ 2 прервали лечение в связи с прогрессированием заболевания. Первичной локализацией опухоли являлась толстая кишка (65%), прямая кишка (29%) или обе (6%). На момент включения в исследование мутации KRAS отмечались у 57% пациентов.
Большинство пациентов (52%) получили 3 или меньше предыдущих курсов противоопухолевой терапии при метастазах. Терапия включала в себя химиотерапию на основе фторпиримидинов, лечение ингибиторами фактора роста эндотелия сосудов (VEGF), и при диком типе KRAS терапию ингибиторами рецептора эпидермального фактора роста (EGFR).
Добавление лекарственного средства Стиварга® к ОПТ показало значительное увеличение выживаемости по сравнению с плацебо плюс ОПТ при значении р равном 0,005178 согласно стратифицированному тесту логарифмических рангов, соотношении рисков равном 0,774 (95% CI 0,636; 0,942) и средней ОВ равной 6,4 месяца по сравнению с 5,0 месяцами. ВБП была значительно больше среди пациентов, принимавших лекарственное средство Стиварга® плюс ОПТ (соотношение рисков HR: 0,494, р<0,000001). Частота ответа (полный ответ или частичный ответ) составила 1% и 0,4% для пациентов принимающих лекарственное средство Стиварга® и плацебо соответственно (р=0.188432). Частота контроля заболевания (полный ответ, частичный ответ или стабильное заболевание) была значительно выше для пациентов, принимающих лекарственное средство Стиварга® (41,0% против 14,9%, р<0,000001).
Анализ подгрупп в отношении общей выживаемости и выживаемости без прогрессирования показал эффективность регорафениба по сравнению с плацебо вне зависимости от возраста (< 65; ≥ 65), пола, статуса по шкале ECOG, первичного очага заболевания, времени от установления диагноза до выявления метастазов, предшествующей противоопухолевой терапии, предшествующих линий терапии метастатического рака и мутации в гене KRAS.
Анализ подгрупп в отношении статуса мутаций в гене KRAS в анамнезе пациентов свидетельствует о том, что у пациентов, имеющих опухоли, содержащие ген KRAS дикого типа, регорафениб оказывал положительное влияние на показатели ОВ в отличие от плацебо, в то время как у пациентов, имеющих опухоли с мутациями в гене KRAS, наблюдался низкий эффект в числовом эквиваленте; положительное влияние на ВБП в группе регорафениба отмечалось независимо от статуса мутаций в гене KRAS. Отношение рисков (95% ДИ) к показателям общей выживаемости составило 0,653 (от 0,476 до 0,895) у пациентов с опухолями, содержащими ген KRAS дикого типа и 0,867 (от 0,670 до 1,123) у пациентов, которые имели опухоли с мутациями в гене KRAS, без каких-либо признаков неоднородности терапевтического эффекта (тест на незначимые взаимодействия). Отношение рисков (95% ДИ) к показателям выживаемости без прогрессирования составило 0,475 (0,362-0,623) у пациентов с опухолями, содержащими ген KRAS дикого типа и 0,525 (0,425-0,649) у пациентов с опухолями, имеющими мутации в гене KRAS.
Клиническую эффективность и безопасность применения лекарственного средства Стиварга® оценивали в ходе международного, многоцентрового, рандомизированного, двойного слепого плацебо-контролируемого исследования (CONCUR) III фазы, у 204 пациентов монголоидной расы (> 90% из Восточной Азии), с метастатическим колоректальным раком, предварительно получавших лечение, у которых наблюдалось прогрессирование заболевания после химиотерапии на основе фторпиримидинов. Только 59,5% пациентов, включенных в исследование CONCUR, предварительно получали VEGF и EGFR терапию. Основной конечной точкой эффективности была общая выживаемость (ОВ). Добавление лекарственного средства Стиварга® к ОПТ привело к достоверному повышению выживаемости пациентов по сравнению с комбинацией плацебо и ОПТ, отношение рисков составило 0,550 (р = 0,000159 по стратифицированному тесту логарифмических рангов), а средняя ОВ составляла 8,8 месяца и 6,3 месяца (95 % ДИ 0,395; 0,765).
Показатель ВВП был значительно выше у пациентов, получавших лекарственное средство Стиварга® и ОПТ (отношение рисков: 0,311, р <0,000001), средняя ВВП составила 3,2 месяца с лекарственным средством Стиварга® и 1,9 месяца с плацебо. Профиль безопасности лекарственного средства Стиварга® плюс ОПТ в исследовании CONCUR был сопоставим с профилем безопасности, полученным в исследовании CORRECT.
Метастатические гастроинтестинальные стромальные опухоли (ГИСО)
Клиническая эффективность и безопасность лекарственного средства Стиварга® была оценена в международном многоцентровом рандомизированном двойном слепом плацебо — контролируемом исследовании III фазы при лечении пациентов с гастроинтестинальными стромальными опухолями (ГИСО), ранее получавших терапию двумя ингибиторами тирозинкиназы (иматинибом и сунитинибом).
Анализ основного показателя эффективности — выживаемости без прогрессирования заболевания (ВВП) — проводили после регистрации 144 случаев ВВП. Также оценивали вторичные показатели, в том числе время до возобновления заболевания (ВДВ) и общую выживаемость (ОВ).
Всего было рандомизировано 199 пациентов с ГИСО в соотношении 2:1, принимавших регорафениб в дозе 160 мг плюс ОПТ или плацебо плюс ОПТ (N=66) перорально один раз в день (N=133) в течение 3 недель, с последующим перерывом в приеме продолжительностью в 1 неделю. Средняя суточная принимаемая доза регорафениба составила 140 мг.
Лечение продолжалось до прогрессирования заболевания или появления клинически неприемлемых признаков токсичности. Пациентам, получавшим плацебо, у которых наблюдалось возобновление заболевания, было предложен открытое применение регорафениба (перекрестный вариант). Пациенты, получавшие регорафениб, у которых наблюдалось возобновление заболевания и для кого, по мнению исследователя, терапия регорафенибом приносила клиническую пользу, имели возможность продолжить прием регорафениба в открытом исследовании.
Среди 199 рандомизированных пациентов, средний возраст которых составил 58 лет, 64% составляли мужчины, 68% пациентов составляли представители европеоидной расы, все пациенты имели показатель общего состояния 0 или 1 на исходном уровне по шкале общего состояния (ОС) ECOG. Общее среднее время с момента последнего возобновления заболевания или рецидива до момента рандомизации составило 6 недель.
ВВП была значительно выше среди пациентов, принимавших регорафениб плюс ОПТ, чем плацебо плюс ОПТ с соотношением рисков 0,268 [95% CI 0,185; 0,388] и средней ВВП 4,8 месяцев по сравнению с 0,9 месяцами (р < 0,000001). Относительный риск возобновления заболевания или смерти снизился примерно на 73,2% у пациентов, принимавших регорафениб, по сравнению с пациентами, принимавшими плацебо. Увеличение ВВП носило постоянный характер независимо от возраста, пола, географического региона, ранее проводимого лечения, ОС по шкале ECOG.
ВДВ было значительно продолжительнее у пациентов, получавших регорафениб плюс ОПТ, чем у пациентов, получавших плацебо плюс ОПТ с соотношением рисков равным 0,248 [95% CI 0,170; 0,364], и среднее ВДВ составило 5,4 месяца по сравнению с 0,9 месяцами (р < 0,000001).
Соотношение рисков для ОВ составило 0,772 (95% CI 0,423; 1,408; р = 0,199); 85% пациентов, изначально рандомизированных в группу плацебо, после возобновления заболевания получали лечение регорафенибом.
Кроме того, 56 пациентов, получавших плацебо плюс ОПТ, в рамках открытого исследования, после перекреста принимали лекарственное средство Стиварга® вследствие возобновления заболевания, и всего 41 пациент, принимавший лекарственное средство Стиварга® плюс ОПТ, продолжили лечение лекарственным средством Стиварга® после возобновления заболевания. Средняя вторичная ВВП (измеренная согласно оценке исследователя) составила 5,0 и 4,5 месяца соответственно.
Печеночно-клеточный рак (ПКР)
Клиническая эффективность и безопасность лекарственного средства Стиварга® была оценена в международном многоцентровом рандомизированном двойном слепом плацебо — контролируемом исследовании III фазы (RESORCE) при лечении пациентов с ПКР, ранее получавших терапию сорафенибом. Первичной конечной точкой оценки эффективности являлась общая выживаемость (ОВ). Вторичными критериями являлись выживаемость без прогрессирования (ВВП), время до возобновления заболевания (ВДВ), уровень объективного ответа опухоли и частота контроля заболевания.
Всего было рандомизировано 573 пациента с ПКР в соотношении 2:1, принимавших регорафениб в дозе 160 мг плюс ОПТ (N=379) или плацебо плюс ОПТ (N=194) перорально один раз в день в течение 3 недель, с последующим перерывом в приеме продолжительностью в 1 неделю. Средняя суточная принимаемая доза регорафениба составила 144 мг.
В исследование включались пациенты, у которых наблюдалось прогрессирование заболевания после терапии сорафенибом, и пациенты с печеночной недостаточностью легкой степени (класс А по шкале Чайлд-Пью). Из исследования исключались пациенты, полностью прекратившие терапию вследствие развития токсичности, вызванной приемом сорафениба, или переносившие менее 400 мг сорафениба один раз в день до отмены терапии. Рандомизация проводилась в течение 10 недель после прекращения терапии сорафенибом. Пациенты продолжали прием лекарственного средства Стиварга® до радиологического или клинического прогрессирования заболевания или выявления признаков неприемлемой токсичности. Однако пациенты могли продолжить терапию лекарственным средством Стиварга® после выявления прогрессирования по усмотрению исследователя.
Для всех 573 рандомизированных пациентов демографические и базовые характеристики заболевания, представленные ниже, были сопоставимы между группами, получавшими терапию лекарственным средством Стиварга® и плацебо:
Медиана возраста: 63 года
Мужчины: 88%
Европеоидная раса:36%, монголоидная раса:41%
ОС по шкале ECOG: 0 баллов — 66%, 1 балл — 34
Печеночная недостаточность: класс А по шкале Чайлд-Пью — 98%, класс В по шкале Чайлд-Пью — 2%
Этиология включала гепатит В (38%), гепатит С (21%), неалкогольный стеатогепатит ASH (7%)
Отсутствие, как макроскопической сосудистой инвазии, так и внепеченочного распространения опухоли: 19%
Рак печени: стадия В — 13% (по BCLC — Барселонская классификация рака печени), стадия С — 87%
Локально-региональная трансартериальная эмболизация или инфузионная химиотерапия: 61%
Лучевая терапия до применения регорафениба: 15%
Медиана продолжительности лечения: 7,8 месяцев
Добавление лекарственного средства Стиварга® к ОПТ показало статистически значимое увеличение выживаемости по сравнению с плацебо плюс ОПТ с соотношением рисков 0,624 (95% CI 0,498; 0,7852), р=0,000017 по стратифицированному тесту логарифмических рангов, и медианой общей выживаемости 10,6 месяцев против 7,8 месяцев.
Фармакокинетика
Абсорбция
Среднее значение пикового уровня регорафениба в плазме крови составляет около 2,5 мг/л приблизительно через 3-4 часа после приема внутрь однократной дозы 160 мг регорафениба — 4 таблетки по 40 мг. При однократной дозе в 60 мг или 100 мг средняя относительная биодоступность таблеток по сравнению с раствором для перорального применения составляет 69% и 83% соответственно.
Наибольшая концентрация регорафениба и его основных фармакологически активных метаболитов (М-2 и М-5) достигается при приеме после завтрака с низким содержанием жиров (легкого) по сравнению с приемом после завтрака с высоким содержанием жиров или приемом натощак. По сравнению с приемом натощак экспозиция регорафениба возрастает на 48% при приеме после завтрака с высоким содержанием жиров и на 36% при приеме после завтрака с низким содержанием жиров. По сравнению с приемом натощак, экспозиция метаболитов М-2 (N — оксид) и М-5 (N — оксид и N — десметил) выше при приеме регорафениба после легкого завтрака с низким содержанием жиров и ниже при приеме после завтрака с высоким содержанием жиров.
Распределение
Кривая зависимости «концентрация — время» демонстрирует несколько пиков как для регорафениба, так и для его основных циркулирующих метаболитов, в течение 24 часов после приема дозы, что связано с печеночно — кишечной рециркуляцией лекарственного средства. In vitro связывание с белками плазмы высокое и составляет 99,5%. In vitro связывание М-2 и М-5 с белками крови выше, чем у регорафениба и составляет 99,8% и 99,95% соответственно. Метаболиты М-2 и М-5 являются слабыми субстратами Р-гликопротеина. Метаболит М-5 является слабым BCRP-субстратом.
Метаболизм
Регорафениб метаболизируется главным образом в печени посредством окислительного метаболизма, опосредованного изоферментом CYP3A4, а также путем глюкуронизации, опосредованной UGT1A9. В плазме были идентифицированы два основных и шесть второстепенных метаболитов регорафениба. Основными циркулирующими метаболитами регорафениба в плазме крови являются М-2 (N-оксид) и М-5 (N-оксид и N-десметил), которые представляют собой фармакологически активные вещества и в равновесном состоянии имеют концентрацию аналогичную регорафенибу. М-2 метаболизируется посредством окислительного метаболизма, опосредованного изоферментом CYP3A4, а также путем глюкуронизации, опосредованной UGT1А9.
Метаболиты могут быть восстановлены или гидролизованы микрофлорой желудочно-кишечного тракта, при этом возможна реабсорбция неконъюгированного активного вещества и его метаболитов (печеночно-кишечная рециркуляция).
Выведение
После перорального приема средний период полувыведения из плазмы для регорафениба и его метаболита М-2 по данным различных исследований составляет от 20 до 30 часов. Средний период полувыведения для метаболита М-5 составляет приблизительно 60 часов (варьируется от 40 до 100 часов).
Приблизительно 90% меченой изотопами дозы выводится в течение 12 дней после приема, около 71% дозы выделяется с калом (47% в виде исходного соединения, 24% в виде метаболитов) и около 19% дозы выделяется с мочой в виде глюкуронидов. В равновесном состоянии выведение глюкуронидов почками уменьшается и составляет менее 10%. Исходное соединение, обнаруженное в кале, может быть продуктом неабсорбированного лекарственного средства, желудочно-кишечного расщепления глюкуронидов либо восстановления метаболита М-2.
М-5 может расщепляться микрофлорой желудочно-кишечного тракта до М-4, обеспечивая реабсорбцию М-4 (печеночно-кишечная рециркуляция). М-5 окончательно выводится с калом как М-6 (карбоновая кислота) через М-4.
Линейность/нелинейность
Системное воздействие регорафениба при равновесной концентрации возрастает пропорционально дозе при применении до 60 мг и менее пропорционально при дозах более 60 мг. Накопление лекарственного средства при равновесной концентрации приблизительно в два раза превышает концентрацию в плазме, которая соответствует периоду полувыведения и частоте приема лекарственного средства. После перорального приема регорафениба в дозе 160 мг его средняя максимальная концентрация в плазме крови в равновесном состоянии достигает 3,9 мг/л (8,1 мкмоль). Соотношение максимальной и минимальной концентрации регорафениба в плазме крови составляет менее 2.
Оба метаболита М-2 и М-5 демонстрируют нелинейный характер накопления, что может быть обусловлено гепато-энтеральной циркуляцией или насыщением UGT1A9 пути. Несмотря на то, что после однократного приема регорафениба концентрации М-2 и М-5 в плазме крови намного ниже, чем у исходного соединения, в равновесном состоянии плазменные концентрации М-2 и М-5 аналогичны концентрациям регорафениба.
Печеночная недостаточность
Воздействие регорафениба и его метаболитов М-2 и М-5 сопоставимо у пациентов с печеночной недостаточностью легкой степени (класс А по шкале Чайлд-Пью) и у пациентов с нормальной печеночной функцией.
Ограниченные данные по пациентам с умеренной печеночной недостаточностью (класс В по шкале Чайлд-Пью) указывают на величину экспозиции, аналогичную той, что наблюдается у пациентов с нормальной печеночной функцией после однократной дозы в 100 мг регорафениба. Отсутствуют какие-либо данные для пациентов с печеночной недостаточностью класса С по шкале Чайлд-Пью (тяжелая степень). Поскольку регорафениб выводится из организма в основном печенью, у пациентов с тяжелыми нарушениями функции печени возможно повышение экспозиции.
Почечная недостаточность
Доступные клинические данные и данные фармакокинетического моделирования, основанные на физиологических показателях, указывают на схожие экспозиции регорафениба и его метаболитов М-2 и М-5 в равновесном состоянии у пациентов с легкой или умеренной степенью почечной недостаточности в сравнении с пациентами с нормальной функцией почек. Фармакокинетика регорафениба не была изучена у пациентов с острой почечной недостаточностью или терминальной стадией почечной недостаточности. Тем не менее, данные фармакокинетического моделирования, основанного на физиологических показателях, не предусматривают релевантных изменений экспозиции у таких пациентов.
Пациенты пожилого возраста
Возраст не оказывает влияния на фармакокинетику регорафениба в пределах изученного возрастного диапазона (29-85 лет).
Пол
Не выявлено различий в фармакокинетике регорафениба в зависимости от пола.
Этническая принадлежность
Не выявлено различий в фармакокинетических параметрах регорафениба в зависимости от этнической принадлежности.
Электрофизиология сердца/удлинение интервала ОТ
У пациентов женского и мужского пола с онкологическими заболеваниями не было выявлено удлинения интервала QT в равновесном состоянии при приеме регорафениба в дозе 160мг.
Доклинические данные о безопасности
Системная токсичность
После повторного введения дозы у мышей, крыс и собак наблюдались нежелательные реакции со стороны ряда органов, прежде всего почек, печени, пищеварительного тракта, щитовидной железы лимфатической/кроветворной системы, эндокринной системы, репродуктивной системы и кожи. В 26-недельном исследовании токсичности при введении повторных доз у крыс наблюдалось небольшое увеличение частоты случаев утолщения атриовентрикулярных клапанов сердца. Причиной данного явления может служить ускорение возрастного физиологического процесса. Данные реакции наблюдались при системных экспозициях, находящихся в диапазоне или ниже диапазона предполагаемой экспозиции у человека (основано на сравнении AUC). Изменения зубов и костей, а также нежелательные явления в репродуктивной системе были наиболее выражены у молодых и растущих животных, а также у молодых крыс, что указывает на потенциальный риск для детей и подростков.
Тератогенность и эмбриотоксичность
Специальные исследования влияния регорафениба на фертильность не проводились. Следует учитывать способность регорафениба оказывать неблагоприятное воздействие на репродуктивную систему мужчин и женщин. В исследовании на крысах и собаках после многократного применения регорафениба при экспозициях ниже предполагаемой экспозиции у человека (при сравнении AUC) наблюдались морфологические изменения в яичках, яичниках и матке. Наблюдаемые изменения были обратимы только частично.
В исследовании на кроликах при экспозиции ниже предполагаемой экспозиции у человека наблюдалась эмбриотоксичность (при сравнении AUC). В основном были обнаружены нарушения формирования мочевыделительной системы, сердца и крупных сосудов.
Генотоксичность и канцерогенность
Регорафениб не показал генотоксичности в исследованиях in vitro и in vivo на мышах.
Исследования канцерогенности регорафениба не проводились.
Общий профиль безопасности препарата Стиварга® оценивается на данных клинических исследований с участием более 1200 пациентов, получавших терапию в плацебо-контролируемых клинических исследованиях III фазы. Данная группа включала 500 пациентов с метастатическим колоректальным раком и 132 пациента с гастроинтестинальными стромальными опухолями.
В клинических исследованиях наиболее частыми нежелательными реакциями (≥30% пациентов) были астения/усталость, ладонно-подошвенная эритродизестезия, диарея, снижение аппетита и потребления пищи, повышение АД, дисфония, инфекции.
Наиболее серьезными нежелательными реакциями при приеме препарата Стиварга® были поражения печени, кровотечения, прободение ЖКТ.
Перечисленные ниже нежелательные явления, отмеченные при применении препарата Стиварга® в ходе клинических исследований, распределены по частоте возникновения в соответствии со следующей градацией: очень часто (≥1/10), часто (от ≥1/100 до <1/10), нечасто (от ≥1/1000 до <1/100), редко (от ≥1/10000 до <1/1000).
Для классификации и описания конкретной реакции, ее синонимов и связанных с ней состояний, используется наиболее подходящий термин из MedDRA.
В каждой частотной группе нежелательные явления представлены в порядке уменьшения их значимости.
Со стороны крови и лимфатической системы: очень часто — тромбоцитопения, анемия; часто — лейкопения.
Со стороны сердца и сосудов: очень часто — кровотечения1, повышение АД; нечасто — инфаркт миокарда, ишемия миокарда, гипертонический криз.
Со стороны дыхательной системы, органов грудной клетки и средостения: очень часто — дисфония.
Со стороны кожи и подкожных тканей: очень часто — ладонно-подошвенная эритродизестезия, кожная сыпь, алопеция; часто — сухость кожи, эксфолиативный дерматит; нечасто — поражение ногтей, мультиформная эритема; редко — синдром Стивенса-Джонсона, токсический эпидермальный некролиз.
Со стороны ЖКТ: очень часто — диарея, стоматит, рвота, тошнота; часто — нарушение вкуса, сухость слизистой оболочки полости рта, гастроэзофагеальный рефлюкс, гастроэнтерит; нечасто — прободение ЖКТ1, свищ ЖКТ.
Со стороны печени и желчевыводяших путей: очень часто — гипербилирубииемия; часто — повышение активности трансаминаз; нечасто — тяжелое нарушение функции печени1,2.
Со стороны нервной системы: очень часто — головная боль; часто — тремор; редко — синдром задней обратимой энцефалопатии.
Со стороны скелетно-мышечной и соединительной ткани: часто — мышечно-скелетная ригидность.
Со стороны почек и мочевыводящих путей: часто — протеинурия.
Со стороны эндокринной системы: часто — гипотиреоз.
Со стороны обмена веществ и питания: очень часто — снижение аппетита и потребления пищи; часто — гипокалиемия, гипофосфатемия, гипокальциемия, гипонатриемия, гипомагниемия, гиперурикемия.
Лабораторные и инструментальные данные: очень часто — снижение массы тела; часто — увеличение активности амилазы и липазы, отклонение от нормального значения MHO.
Доброкачественные, злокачественные и неуточненные новообразования (включая кисты и полипы): редко — кератоакантома/плоскоклеточный рак кожи.
Инфекционные и паразитарные заболевания: очень часто — инфекции.
Общие расстройства и нарушения в месте введения: очень часто — астения/общая слабость, боль различной локализации, повышение температуры тела, воспаление слизистых оболочек.
1 Сообщалось о летальном исходе в результате неблагоприятной реакции.
2 В соответствии с критериями Международной рабочей группы экспертов по лекарственному поражению печени.
Поражения печени. Тяжелое лекарственное поражение печени со смертельным исходом наблюдалось у трех пациентов из более чем 1200 пациентов, принимавших участие во всех клинических исследованиях и получавших терапию препаратом Стиварга® (0,25%). У двоих из данных пациентов отмечались метастазы в печени. Дисфункция печени у данных пациентов началась в течение первых двух месяцев терапии и характеризовалась повреждением гепатоцитов с повышением активности трансаминаз >20×ВГН, сопровождаемым увеличением концентрации билирубина. В результате биопсии печени у двух пациентов был обнаружен некроз клеток печени с воспалительным инфильтратом.
Кровотечения. В двух плацебо-контролируемых исследованиях III фазы общая частота кровотечений у пациентов, получавших препарат Стиварга®, составила 19,3%. Большинство случаев кровотечения имели легкую или умеренную степень тяжести (1-я и 2-я степень: 16,9%). Наиболее часто отмечалось носовое кровотечение (7,6%).
Летальные исходы у пациентов, получавших препарат Стиварга®, отмечались редко (0,6%) и чаще были связаны с поражением дыхательной, пищеварительной и мочеполовой систем.
Инфекции. В двух плацебо-контролируемых исследованиях III фазы инфекционные заболевания чаще отмечались у пациентов, получавших лечение препаратом Стиварга®, по сравнению с пациентами, получавшими плацебо (все степени: 31% по сравнению с 14,4%). Чаще инфекции у пациентов, получавших препарат Стиварга®, имели легкую или умеренную степень выраженности (1-я и 2-я степень: 22,9%) и включали инфекции мочевыводящих путей (6,8%), назофарингит (4,2%), а также кандидоз кожи и слизистых и системный микоз (2,4%). Не наблюдались различия по частоте летальных исходов в связи с развитием инфекции у пациентов, получавших препарат Стиварга® (0,6%) и у пациентов, получавших плацебо (0,6%).
Ладонно-подошвенная эритродизестезия. В плацебо-контролируемом исследовании III фазы общая частота возникновения ладонно-подошвенной эритродизестезии у пациентов с метастатическим колоректальным раком, принимавших препарат Стиварга®, составила 45,2% и у пациентов, получающих плацебо — 7,1%. В плацебо-контролируемом исследовании III фазы у пациентов с гастроинтестинальными стромальными опухолями общая частота возникновения ладонно-подошвенной эритродизестезии составила 66,7% — у пациентов, получавших терапию препаратом Стиварга®, и 15,2% — у пациентов, получавших плацебо. В обоих исследованиях большинство случаев ладонно-подошвенной эритродизестезии у пациентов, получавших препарат Стиварга® отмечалось во время первого цикла лечения и имело легкую или среднюю степень тяжести (1-я и 2-я степени: 28,6% у пациентов — с метастатическим колоректальным раком и 44,7 % — у пациентов с гастроинтестинальными стромальными опухолями). Частота возникновения ладонно-подошвенной эритродизестезии 3-й степени составила 16,6% у пациентов с метастатическим колоректальным раком и 22% у пациентов с гастроинтестинальными стромальными опухолями.
Повышение АД. В плацебо-контролируемом исследовании III фазы у пациентов с метастатическим колоректальным раком общая частота случаев повышения АД составила 30,4% у пациентов, принимавших препарат Стиварга®, и 7,9% у пациентов, принимавших плацебо. В плацебо-контролируемом исследовании Ш фазы у пациентов с гастроинтестинальными стромальными опухолями общая частота случаев повышения АД составила 59,1% — у пациентов, получавших препарат Стиварга®, и 27,3% — у пациентов, получавших плацебо. В обоих исследованиях большинство случаев повышения АД у пациентов, получавших препарат Стиварга®, было зарегистрировано в течение первого цикла лечения. При этом случаи повышения АД имели легкую и умеренную степень тяжести (1-я и 2-я степень: 22,8% — у пациентов с метастатическим колоректальным раком и 31,1% — у пациентов с гастроинтестинальными стромальными опухолями). Частота случаев повышения АД 2-й степени составила 7,6% (у пациентов с колоректальным раком) и 27,3% (у пациентов с гастроинтестинальными стромальными опухолями). Был зарегистрирован один случай повышения АД 4-й степени у пациента с гастроинтестинальной стромальной опухолью.
Протеинурия. В плацебо-контролируемом исследовании III фазы среди пациентов с метастатическим колоректальным раком частота случаев протеинурии, связанной с лечением, составила 7,4% у пациентов, получавших терапию препаратом Стиварга®, в сравнении с 2,4% у пациентов, принимавших плацебо. После развития протеинурии у 40,5% пациентов из группы препарата Стиварга® и 66,7% пациентов из группы плацебо состояние не вернулось к исходным значением. В плацебо-контролируемом исследовании III фазы среди пациентов с гастроинтестинальными стромальными опухолями общая частота случаев протеинурии составила 6,8% у пациентов, получающих терапию препаратом Стиварга®, в сравнении с 1,5% у пациентов, принимавших плацебо.
Со стороны сердца и сосудов. Во всех проводимых клинических исследованиях нежелательные явления в виде нарушений со стороны сердца (все степени) чаще регистрировались (20,5 относительно 10,4%) у пациентов, получавших терапию препаратом Стиварга®, в возрасте 75 лет или старше (N=78), чем у пациентов, получавших терапию препаратом Стиварга®, в возрасте до 75 лет (N=995).
Лабораторные и инструментальные данные. В двух плацебо-контролируемых исследованиях III фазы у 26,1% пациентов, получавших терапию препаратом Стиварга®, и у 15,1% пациентов, получавших плацебо, концентрация ТТГ была выше ВГН. Значения ТТГ в 4 раза превышающие ВГН были зарегистрированы у 6,9% пациентов, получавших терапию препаратом Стиварга®, и у 0,7% пациентов, принимавших плацебо. Концентрация свободного трийодтиронина (Т3 свободный) меньше нижней границы нормы отмечалась у 25,6% пациентов, получающих терапию Стиварга®, и у 20,9% пациентов, получающих плацебо. Концентрация свободного тироксина (Т4 свободный) меньше нижней границы нормы отмечалась у 8% пациентов в группе терапии препаратом Стиварга® и у 6,6% пациентов из группы плацебо. В целом, приблизительно у 7% пациентов, получающих терапию препаратом Стиварга®, развился гипотиреоз, требующий применения заместительной гормонотерапии.