Ожирение и избыточный вес ведут к развитию многочисленных серьезных осложнений. В настоящее время разработан и находится в завершающей стадии исследований новый препарат для снижения веса – тезофензин (tesofensine), который модулирует синаптический обратный захват моноаминов, снижая частоту приема пищи и повышая затраты энергии. Тезофензин показал эффективность при ожирении, в том числе при диабете типа 2 и синдроме Прадера-Вилли. Препарат активен при пероральном применении и в небольших дозировках — от 0,5 мг. В настоящем обзоре рассмотрены вопросы безопасности и эффективности препарата, согласно данным клинических испытаний.
Ожирение
Ожирение давно превратилось в глобальную эпидемию, которая стимулирует возникновение множества сопутствующих заболеваний: сахарный диабет типа 2, дислипидемия, инсулинорезистентность, сердечно-сосудистые заболевания (инсульт и ишемия), различные типы рака (груди, почек, желчного пузыря, толстой кишки, простаты) и нервно-психические расстройства [1]. Эти сопутствующие заболевания приводят к инвалидности и сокращению продолжительности жизни. Ожирение характеризуется повышенным аномальным накоплением жира, которое может быть результатом дисбаланса энергетического гомеостаза, то есть повышенного потребления и снижения расхода энергии (например, из-за отсутствия физической активности). К основным этиопатологическим факторам ожирения относятся генетическая предрасположенность, малоподвижный образ жизни с недостаточным расходом энергии и факторы окружающей среды, например, потребление высококалорийной пищи. Во всем мире около 2,8 миллиона человек ежегодно умирают из-за наличия избыточного веса и/или ожирения [2]. Например, распространенность ожирения в США к 2018 году составляла 42,4% с поправкой на возраст, а тяжелого ожирения – 9,2% среди лиц в возрасте >20 лет. Средняя распространенность была одинаковой среди представителей обоих полов, но случи тяжелого были выше среди женщин. Среди лиц среднего возраста (40–59 лет) тяжелое ожирение имеет самую высокую распространенность [3].
Стратегии лечения ожирения включают изменение образа жизни, бариатрическую хирургию и фармакотерапию [4]. Доступные сейчас препараты от ожирения, одобренные Управлением по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA), включают фентермин, орлистат, лорказерин и лираглутид. Из-за сложной и многофакторной патофизиологии ожирения, монотерапия не является адекватной, поэтому зачастую применяются многоцелевые подходы для повышения терапевтической пользы. Комбинации, одобренные FDA, представляют собой препараты с замедленным высвобождением налтрексона/бупропиона и составы с пролонгированным высвобождением фентермина/топирамата [4]. Многие препараты, такие как тезофензин, комбинация бупропион/зонисамид, эксенатид, цетилистат, RM-493 (сетмеланотид), KD-026 и ремоглифлозина этабонат, находятся на разных этапах клинических исследований. Текущие разработки лекарств для лечения ожирения ориентированы на воздействие на центральные пути. Эти препараты, в основном, уменьшают потребление пищи и увеличивают расход энергии. Одним из новых потенциальных лекарственных средств такого типа, все еще находящемся на стадии клинически исследований, является тезофензин.
Тезофензин
Тезофензин (Tesofensine, NS-2330; (1R,2R,3S,5S)-3-(3,4-дихлорфенил)- 8-метил-2-(этоксиметил)-8-азабицикло[3.2.1]октан; CAS 195875-84-4) – это производное азабициклооктана, представляющее собой новое лекарство от ожирения, которое действует центрально путем тройного ингибирования обратного захвата моноаминов (Рис. 1).

Рисунок 1 – Структурная формула тезофензина
Тезофензин модулирует синаптический обратный захват дофамина (DA), норэпинефрина (NE) и серотонина (5-HT) в синаптической щели [5]. Эти моноамины принимают ключевое участие в регуляции аппетита, расходе энергии и контроле массы тела. Тезофензин был первоначально разработан для лечения нейродегенеративных заболеваний, таких как болезни Альцгеймера и Паркинсона [6-8]. Эффективность лечения данных заболеваний оказалась невысока [7, 9], но при этом сообщалось, что во время клинических исследований тезофензин вызывает серьезную потерю веса у пациентов, участвовавших в эксперименте, особенно у страдающих наличием избыточного веса или ожирения [10].
Результаты наблюдений подтолкнули исследователей к разработке тезофензина в качестве нового терапевтического агента для лечения ожирения, направленного на центральный обратный захват моноаминов и холинергическую нейротрансмиссию. Кроме того, исследование показало, что повышенная регуляция дофаминергических путей у субъектов, получавших тезофензин, оказалась следствием блокады DAT (переносчика дофамина) и последующего увеличения концентрации дофамина [11].
Тезофензин первоначально был разработан и запатентован компанией NeuroSearch A/S (Дания) для лечения болезни Паркинсона и болезни Альцгеймера. Первый синтез препарата описан в патенте [12], и, затем был усовершенствован [13]. В настоящее время работу над тезофензином ведут Saniona (Дания) в партнерстве с Productos Medix S.A. de C.V. (Мексика) [14].
Изначально тезофензин показал серьезную потерю веса в ходе исследования фазы I, и, следовательно, его перепрофилировали для лечения ожирения. Исследование фазы II показало, что даже доза тезофензина 0,5 мг в течение 24 недель способна вызвать значительную потерю веса. У умеренно здоровых мужчин тезофензин приводил к существенной потере веса за счет снижения аппетита и увеличения расхода энергии по сравнению с плацебо. В доклинических исследованиях было обнаружено, что тезофензин действует центрально, подавляя аппетит, регулируя доступность дофаминовых рецепторов и стимулируя α1-адренорецепторы. Также наблюдалась стимулирующая дофаминергическая реакция тезофензина в прилежащем ядре и префронтальной коре головного мозга. Кроме того, тезофензин изменяет экспрессию дофаминергических рецепторов в полосатом теле мозга, а также их связывание. Активная регуляция дофаминергического пути наблюдалась в результате увеличения синаптической доступности дофамина из-за дозозависимой блокады переносчика дофамина. Препарат имеет длительный период полувыведения, который составляет 8–9 дней, и в основном метаболизируется в печени с помощью CYP3A4 до дезалкильного метаболита M1 (NS-2360), который является единственным метаболитом, обнаруживаемым в плазме крови человека [2]. Далее в обзоре рассматриваются фармакологические аспекты тезофензина как потенциального средства против ожирения.
Доклинические и клинические исследования
При изучении терапии болезни Паркинсона было обнаружено, что тезофензин ингибирует обратный захват моноаминов DA, NE и 5-HT наряду со стимуляцией холинергических нейронов в префронтальной коре и гиппокампе [15].
Для изучения фармакодинамики вызванного тезофензином подавления аппетита, крысам с индуцированным диетой ожирением вводили тезофензин по 2 мг/кг подкожно в течение 16 дней [16]. Препарат значительно, до -25%, снизил массу тела животных, демонстрируя гипофагические эффекты с ED50 1,3 мг/кг. Высокие доза препарата (2 и 3 мг/кг) уменьшали общую массу потребляемой крысами пищи в 2,5–3 раза. Гипофагический ответ на тезофензин был отменен совместным введением антагониста α1-адренорецепторов, празозина, (1 мг/кг), и частично антагонизирован SCH-23390 (0,03 мг/кг). Напротив, антагонисты рецепторов D2, D3, α2- и 5-HT2A/C не влияли на гипофагию. Таким образом, было доказано, что тезофензин вызывает гипофагию за счет стимуляции рецепторов α1 и D1. В другом исследовании показали, что тезофензин столь же эффективен на грызунах, как и пероральный прием сибутрамина в дозировке 7,5 мг/кг [17].
На модели болезни Паркинсона, вызванной с помощью нейротоксичного МФТП у мартышек, он показал уменьшение проявлений симптомов Паркинсона без вызывания дискинезии. И наоборот, тезофензин сам по себе или в комбинации с леводопой не излечивал симптомы у пациентов с запущенной болезнью Паркинсона [6], а также у пациентов с ранней стадией заболевания [7]. В 2004 году исследования показали, что тезофензин умеренно улучшает прогрессирующую стадию болезни Паркинсона, но зависимости от дозы не было установлено. Более 20% пациентов ответили на терапию тезофензином и показали улучшение по подшкале II UPDRS (унифицированной рейтинговой шкале болезни Паркинсона) и общий балл по подшкале III. Тезофензин (0,5 мг) продемонстрировал статистически значимое улучшение показателей UPDRS по сравнению с плацебо. Однако умеренное улучшение симптомов, отсутствие зависимости доза-ответ и возникновение дискинезии были причиной неудачи и прекращения изучения тезофензина в качестве лекарственного средства для лечения болезни Паркинсона [9].
При болезни Альцгеймера тезофензин усиливал функцию основных нейротрансмиттеров, таким образом косвенно стимулируя холинергическую систему. Ацетилхолин играет ключевую роль в патофизиологии болезни Альцгеймера. Было обнаружено, что тесофенсин снижает уровень β-амилоида у мышей и обладает нейропротекторным действием. Тезофензин показал значительное улучшение когнитивных функций у пациентов с болезнью Альцгеймера в клинических испытаниях фазы IIa, но фаза IIb показала скомпрометированные результаты, и дальнейшие исследования были прекращены в 2008 году [18]. Исследования in vitro показали, что его метаболит (M1) имеет больший потенциал, чем исходный препарат. Исследования in vivo, проведенные на мышах в диапазоне дозировок тезофензина 0,3–20 мг/кг, также показали, что значение ЕС50 для M1 как ингибитора DAT в 4–6 раз выше [5]. Хотя препарат не проявил значительной эффективности при первоначальной терапии болезней Паркинсона и Альцгеймера, он действительно вызывал значительную потерю веса у пациентов [10].
Поскольку дефицит моноаминов в головном мозге является ключевым признаком депрессии, ингибиторы обратного захвата моноаминов, такие как 5-HT, NE и DA, могут быть полезны для поддержания нормальной физиологии мозга. Но тезофензин не исследовался ни доклинически, ни клинически на предмет наличия антидепрессивных свойств. На животных моделях было показано, что тесофенсин обладает нейропротекторной активностью за счет увеличения уровней нейротрофических факторов головного мозга, а также увеличения пролиферации нейронов гиппокампа крыс [8]. Это исследование продемонстрировало возможное значение тезофензина как нового фармакотерапевтического средства для лечения депрессии [19].
При ожирении тезофензин вызывал заметную потерю веса в исследовании фазы II, проведенном при изучении лечения болезней Альцгеймера и Паркинсона. Это открытие изменило весь процесс разработки лекарственного препарата. В метаанализе сравнили четыре исследования, в которых сообщалось о потере веса у пациентов с болезнью Альцгеймера и Паркинсона, получавших тезофензин. Результаты, полученные на 740 пациентах, показали, что 14-недельное лечение тезофензином (0,125, 0,25, 0,5 и 1,0 мг) приводило к потере веса дозозависимым образом (-0,5%, -0,9%, -1,8% и -2,8%, соответственно) по сравнению с плацебо (+0,5%) [10]. Анализ подгруппы с ожирением показал потерю веса ≥5% у 32% пациентов без применения диеты или изменения образа жизни. Также наблюдали изменение частоты сердечных сокращений (4–7 ударов в минуту) в зависимости от дозы. Похудание было связано с гипофагией (сокращение потребления пищи) и повышенным расходом энергии [10, 16]. Были необходимы дальнейшие исследования, чтобы изучить молекулярный механизм действия препарата против ожирения.
Для изучения механизм действия тезофензина на анорексигенный эффект, снижение веса и расход энергии у людей с избыточным весом или ожирением 32 здоровых добровольца получали ежедневно в течение 7 дней по 2 мг тезофензина [20]. Тезофензин вызвал потерю веса в среднем на 1,8 кг по сравнению с плацебо, наряду с пониженным потреблением пищи. Также наблюдались более высокие затраты энергии в течение ночи и увеличенное суточное окисление жиров по сравнению с плацебо.
Предпочтительный путь введения тезофензина – пероральный, поскольку он очень хорошо всасывается в желудочно-кишечном тракте. Стабильная концентрация в плазме может быть достигнута через 4–6 недель лечения тезофензином. Концентрация лекарственного средства в плазме увеличивалась по логарифмически-линейной зависимости от введенной дозы. Фармакокинетический профиль препарата является линейным после однократного и многократного приема во всех испытанных диапазонах доз, а расчетная абсолютная биодоступность после перорального приема превышает 90% [15]. Концентрация в плазме увеличивается с дозой, но четкой зависимости доза-ответ не наблюдалось. Для достижения максимальной концентрации в плазме (Cmax) требуется 6–8 часов. Наивысший достигнутый объем распределения (Vd) составил 600 л после внутривенного введения. Тезофензин метаболизируется преимущественно до соответствующего дезалкильного метаболита M1 ферментом CYP3A4. Тезофензин и М1 имеют длительный период полураспада, составляющий примерно 200 и 400 часов у человека, соответственно [6]. M1 является единственным обнаруживаемым метаболитом в организме человека, который имеет такие же фармакологические эффекты, как и исходное соединение, тогда как у мышей его активность в 5 раз меньше. Имея период полувыведения около 8 дней у людей, тезофензин может повышать концентрацию дофамина в полосатом теле без фазовых колебаний [9]. Клиренс креатинина был разным у мужчин и женщин: относительный общий клиренс около 1,92 л/ч у мужчин и 1,56 л/ч у женщин, последнее значение на 18,7% меньше, чем у мужчин [18].
Исследование фазы IIа (NCT03149445) представляло собой эксперимент, состоящий из двух частей, в котором проверялось терапевтическое воздействие перорального приема препарата Tesomet [комбинация фиксированных доз тезофензина и метопролола (син. вазокардин, корвитол, метолол)]. В первой части этого исследования – предварительные результаты были опубликованы в 2018 году – шесть взрослых с редким наследственным заболеванием, синдромом Прадера-Вилли (PWS), одним из симптомов которого является ожирение, ежедневно в течение трех месяцев применяли Tesomet (0,5 мг тезофензина и 50 мг метопролола), в то время как трое других пациентов получали плацебо. Результаты показали, что у тех, кто получал Tesomet, наблюдалась клинически значимая потеря веса и уменьшение чрезмерного переедания (гиперфагия). Изменение массы тела составило 6,76% в группе Tesomet по сравнению с 0,75% в группе плацебо; средняя окружность талии уменьшилась на 10 см у пациентов, получавших Tesomet, по сравнению с 6,5 см в группе плацебо. Уменьшение гиперфагии (аномально повышенного аппетита) также наблюдалось в конце исследования. Фактически, после одной недели лечения общий балл гиперфагии упал с 10 на исходном уровне до 5,67 в группе, принимавшей Tesomet, что эквивалентно снижению на 43%. Во второй части Tesomet (тезофензин 0,125 мг плюс метопролол 25 мг) исследовали на взрослых и подростках с PWS. Компания Saniona сообщает, что пациенты показали уменьшение веса, индекса массы тела (ИМТ) и показателя гиперфагии, измеряющего снижение аппетита, при лечении препаратом Tesomet в течение 24-недельного курса лечения [21]. Более высокая доза Tesomet (0,25 мг/день) оказала положительное влияние на массу тела и ИМТ у всех трех подростков с PWS по сравнению с плацебо и дозой 0,125 мг/день. В течение трех месяцев средняя масса тела трех пациентов снизилась на 2,6%, а их средний ИМТ снизился на 4,0%. Средний исходный балл для трех пациентов в начале исследования составлял 8,7. После 3-месячного периода приема более высокой дозы (0,25 мг/день Tesomet) средний балл составил 2,7, что на 69% меньше по сравнению с исходным уровнем. Отметим, что сочетание тезофензин и метопролол изучали в исследованиях NCT03488719 и NCT02737891.
На 60 пациентах с диабетом типа 2 была проведена оценка безопасности и эффективности совместного назначения тесофензина и метопролола (NCT02737891). Исследуемый препарат вводили в течение девяноста дней. Субъекты были случайным образом распределены в одну из двух групп (1:1), которым назначили Tesomet (0,5 мг тезофензина и 100 мг метопролола) или плацебо. В группе, получавшей препарат, средняя масса теkа снизилась на 3,5 кг против 0,3 кг в группе плацебо. У 3% пациентов отмечали сердцебиение и тахикардию.
На основании предыдущих исследований было установлено, что тезофензин в целом безопасен и эффективен, что подтверждается продолжающимися исследованиями. Однако у нескольких пациентов, получавших тезофензин, наблюдалась дискинезия, головная боль, тошнота, запор, галлюцинации и бессонница. Все сообщенные нежелательные явления были оценены как легкие. По результатам метаанализа, частота нежелательных явлений составила 20,4% в группе плацебо и 16,6% в объединенных группах лечения тезофензином [2]. Дискинезия является наиболее частым серьезным нежелательным явлением. В исследовании безопасности наблюдалось дозозависимое увеличение частоты сердечных сокращений. Среднее изменение частоты сердечных сокращений было следующим: плацебо: -0,8 ударов в минуту; группа 0,125 мг тезофензина: 4,7 ударов в минуту; 0,25 мг: 4,7 ударов в минуту; 0,5 мг: 5,6 ударов в минуту; и группа 1 мг: 6,7 ударов в минуту. Среднее изменение систолического артериального давления также было минимальным в группах лечения тезофензином (от -0,29 мм рт.ст. в группе 0,125 мг до -1,95 мм рт.ст. в группе 0,5 мг) по сравнению с небольшим увеличением (0,75 мм рт.ст.) в группе плацебо. Комбинация с метопрололом (Tesomet) разрабатывается для преодоления возникновения сердечных осложнений. Потеря веса наблюдалась у пациентов во всех группах лечения (-0,2 кг в группе плацебо, -0,3 кг в группе 0,125 мг, -0,7 кг в группе 0,25 мг, -0,6 кг в группе 0,5 мг и -1,1 кг в группе 1 мг) [7, 9]. Продолжающиеся в настоящее время клинические испытания могут дать больше информации о аспектах его безопасности. На сегодняшний день лекарственных взаимодействий с тезофензином не наблюдалось [2].
Заключение
Тезофензин – потенциально новое лекарство для похудания, которое в настоящее время проходит III фазу клинических исследований. Он продемонстрировал относительную безопасность и высокую эффективность в нескольких клинических испытаниях фазы I и II. Таким образом, тезофензин представляет собой средство для лечения пациентов с ожирением различной этиологии, включая диабет типа 2 и синдром Прадера-Вилли. Недавно компания Medix подала новую заявку в Управление по контролю за продуктами и лекарствами Мексики (Comisión Federal para la Protección contra Riesgos Sanitarios, COFEPRIS) с целью получения разрешения на применение тезофензина для лечения ожирения [22].

Лоркасерин: снижение аппетита и лечение ожирения
Ожирение и сопутствующие заболевания – серьезная медицинская проблема. Препарат лоркасерин помогает в снижение массы тела, подавлении расстройств пищевого поведения и потенциально полезен для борьбы с…
1. Obesity and overweight. World Health Organization. 2020; Available from: https://www.who.int/en/news-room/fact-sheets/detail/obesity-and-overweight.
2. Singh, R., Kumar, B., Kuhad, A., Tesofensine. Triple monoamine reuptake inhibitor of dopamine, norepinephrine and serotonin; Treatment of obesity. Drugs of the Future, 2018. 43(11): p. 809-814.
3. Prevalence of obesity and severe obesity among adults : United States, 2017–2018, H. National Center for Health Statistics . Division of, Nutrition Examination, S., Editors. 2020: Hyattsville, MD.
4. Srivastava, G., Apovian, C.M., Current pharmacotherapy for obesity. Nature Reviews Endocrinology, 2018. 14(1): p. 12-24. DOI: 10.1038/nrendo.2017.122.
5. Lehr, T., Staab, A., Tillmann, C., Nielsen, E.Ø., Trommeshauser, D., Schaefer, H.G., Kloft, C., Contribution of the active metabolite M1 to the pharmacological activity of tesofensine in vivo: a pharmacokinetic-pharmacodynamic modelling approach. British Journal of Pharmacology, 2008. 153(1): p. 164-174. DOI: 10.1038/sj.bjp.0707539.
6. Bara-Jimenez, W., Dimitrova, T., Sherzai, A., Favit, A., Mouradian, M.M., Chase, T.N., Effect of monoamine reuptake inhibitor NS 2330 in advanced Parkinson’s disease. Movement Disorders, 2004. 19(10): p. 1183-1186. DOI: 10.1002/mds.20124.
7. Hauser, R.A., Salin, L., Juhel, N., Konyago, V.L., Randomized trial of the triple monoamine reuptake inhibitor NS 2330 (tesofensine) in early Parkinson’s disease. Movement Disorders, 2007. 22(3): p. 359-365. DOI: 10.1002/mds.21258.
8. Larsen, M.H., Rosenbrock, H., Sams-Dodd, F., Mikkelsen, J.D., Expression of brain derived neurotrophic factor, activity-regulated cytoskeleton protein mRNA, and enhancement of adult hippocampal neurogenesis in rats after sub-chronic and chronic treatment with the triple monoamine re-uptake inhibitor tesofensine. European Journal of Pharmacology, 2007. 555(2): p. 115-121. DOI: https://doi.org/10.1016/j.ejphar.2006.10.029.
9. Rascol, O., Poewe, W., Lees, A., Aristin, M., Salin, L., Juhel, N., Waldhauser, L., Schindler, T., Group, A.S., Tesofensine (NS 2330), a Monoamine Reuptake Inhibitor, in Patients With Advanced Parkinson Disease and Motor Fluctuations: The ADVANS Study. Archives of Neurology, 2008. 65(5): p. 577-583. DOI: 10.1001/archneur.65.5.577.
10. Astrup, A., Meier, D.H., Mikkelsen, B.O., Villumsen, J.S., Larsen, T.M., Weight Loss Produced by Tesofensine in Patients With Parkinson’s or Alzheimer’s Disease. Obesity, 2008. 16(6): p. 1363-1369. DOI: 10.1038/oby.2008.56.
11. Appel, L., Bergström, M., Buus Lassen, J., Långström, B., Tesofensine, a novel triple monoamine re-uptake inhibitor with anti-obesity effects: Dopamine transporter occupancy as measured by PET. European Neuropsychopharmacology, 2014. 24(2): p. 251-261. DOI: https://doi.org/10.1016/j.euroneuro.2013.10.007.
12. WO 1997030997 A1, 1997.
13. US 7544802 B2, 2009.
14. About Saniona. 2020; Available from: https://saniona.com/about-saniona/.
15. Thatte, U., NS-2330 (Neurosearch). Current opinion in investigational drugs, 2001. 2(11): p. 1592-1594.
16. Axel, A.M.D., Mikkelsen, J.D., Hansen, H.H., Tesofensine, a Novel Triple Monoamine Reuptake Inhibitor, Induces Appetite Suppression by Indirect Stimulation of α1 Adrenoceptor and Dopamine D1 Receptor Pathways in the Diet-Induced Obese Rat. Neuropsychopharmacology, 2010. 35(7): p. 1464-1476. DOI: 10.1038/npp.2010.16.
17. Hansen, H.H., Hansen, G., Tang-Christensen, M., Larsen, P.J., Axel, A.M.D., Raben, A., Mikkelsen, J.D., The novel triple monoamine reuptake inhibitor tesofensine induces sustained weight loss and improves glycemic control in the diet-induced obese rat: Comparison to sibutramine and rimonabant. European Journal of Pharmacology, 2010. 636(1): p. 88-95. DOI: https://doi.org/10.1016/j.ejphar.2010.03.026.
18. Lehr, T., Staab, A., Tillmann, C., Trommeshauser, D., Raschig, A., Schaefer, H.G., Kloft, C., Population pharmacokinetic modelling of NS2330 (tesofensine) and its major metabolite in patients with Alzheimer’s disease. British Journal of Clinical Pharmacology, 2007. 64(1): p. 36-48. DOI: 10.1111/j.1365-2125.2007.02855.x.
19. Marks, D.M., Pae, C.-U., Patkar, A.A., Triple Reuptake Inhibitors: The Next Generation of Antidepressants. Current Neuropharmacology, 2008. 6(4): p. 338-343.
20. Sjödin, A., Gasteyger, C., Nielsen, A.L., Raben, A., Mikkelsen, J.D., Jensen, J.K.S., Meier, D., Astrup, A., The effect of the triple monoamine reuptake inhibitor tesofensine on energy metabolism and appetite in overweight and moderately obese men. International Journal of Obesity, 2010. 34(11): p. 1634-1643. DOI: 10.1038/ijo.2010.87.
21. Higher Dose of Tesomet Effective in PWS Adolescents, Saniona Reports. 2019; Available from: https://praderwillinews.com/2019/09/24/saniona-reports-higher-dose-tesomet-effective-adolescent-pws-patients/.
22. Medix Seeks Approval of Tesofensine to Treat Obesity in Mexico. 2020; Available from: https://praderwillinews.com/2020/01/02/medix-seeks-approval-tesofensine-obesity-mexico/.
 |
|
| Клинические данные | |
|---|---|
| Беременность. категория |
|
| Способы введения. | Оральный |
| Код ATC |
|
| Правовой статус | |
| Правовой статус |
|
| Фармакокинетические данные данные | |
| Биодоступность | 90% |
| Метаболизм | 15–20% почек; печеночный: CYP3A4 |
| Период полувыведения | 220 часов |
| Выведение | Неприменимо |
| Идентификаторы | |
Название IUPAC
|
|
| Номер CAS |
|
| PubChem CID |
|
| ChemSpider |
|
| UNII |
|
| CompTox Dashboard (EPA ) |
|
| Клинические данные | |
| Беременность. категория |
|
| Способы введения. | Оральный |
| Код ATC |
|
| Правовой статус | |
| Правовой статус |
|
| Фармакокинетические данные данные | |
| Биодоступность | 90% |
| Метаболизм | 15–20% почек; печеночный: CYP3A4 |
| Период полувыведения | 220 часов |
| Выведение | Неприменимо |
| Идентификаторы | |
Название IUPAC
|
|
| Номер CAS |
|
| PubChem CID |
|
| ChemSpider |
|
| UNII |
|
| CompTox Dashboard (EPA ) |
|
| Химические и физические данные | |
| Формула | C17H23Cl2NO |
| Молярная масса | 328,28 г · моль |
| 3D-модель (JSmol ) |
|
УЛЫБКИ
|
|
InChI
|
|
| (что это?) |
Тезофенсин (NS2330 ) представляет собой ингибитор обратного захвата серотонина, норадреналина и дофамина из семейства препаратов фенилтропан, который разрабатывается для лечения ожирения. Офенсин был первоначально разработан датской биотехнологической компанией NeuroSearch, которая передала права компании Saniona в 2014 году.
С 2019 года тезофенсин был прекращен для лечения болезней Альцгеймера и Паркинсона, но находится в стадии III клиническое исследование ожирения.
Содержание
- 1 История
- 2 Фармакология
- 2.1 Метаболизм и период полувыведения
- 2.2 Селективность переносчика
- 3 Клинические испытания
- 4 Неблагоприятные события
- 5 Источники
История
Тезофенсин первоначально исследовался для лечения болезни Альцгеймера и болезни Паркинсона, и впоследствии отказались от разработки для этих приложений после того, как первые результаты испытаний показали ограниченную эффективность для лечения этих заболеваний. Однако в исходных исследованиях потеря веса постоянно указывалась как нежелательное явление, особенно у пациентов с избыточным весом или ожирением. Поэтому было решено продолжить разработку тезофензина для лечения ожирения.
Тезофенсин в первую очередь действует как подавитель аппетита, но, возможно, также действует, увеличивая расход энергии в состоянии покоя. Успешно завершены клинические испытания фазы II для лечения ожирения.
Фармакология
Метаболизм и период полувыведения
Тезофензин имеет длительный период полувыведения около 9 дней (220 часов) »и в основном метаболизируется цитохромом P4503A4 (CYP3A4 ) к его дезалкильному метаболиту M1 «NS2360. NS2360 — единственный метаболит, обнаруживаемый в плазме крови человека.
NS2330 в основном метаболизируется цитохромом P450 3A4 (CYP3A4) в.
У него более длительный период полураспада, чем у тезофензина, то есть примерно 16 дней (374 ч) у человека, и его воздействие составляет 31–34% от исходного соединения в устойчивом состоянии. Данные in vivo показывают, что NS2360 отвечает примерно за 6% активности тезофенсина. Как и у животных, почки, по-видимому, играют лишь незначительную роль в клиренсе тезофенсина у людей (около 15–20%).
Селективность транспортера
Первоначально сообщалось, что Тезофенсин имеет IC50 8,0, 3,2 и 11,0 нМ на DAT, NAT и 5HTT. Однако совсем недавно были представлены следующие данные: IC 50 (нМ) NE 1.7, SER 11, DA 65. [цитируется в] Пересмотренные IC 50 адекватно объясняют отсутствие эффективности в лечении болезни Паркинсона,, , т.е. недостаточная эффективность DRI относительно SERT и NET. Это также может помочь объяснить, почему люди, злоупотребляющие стимуляторами, не могут самостоятельно вводить тестофенсин, поскольку считается, что для этого необходимо ингибирование DAT, а не ингибирование NET.
Тезофенсин также косвенно потенцирует холинергическая нейротрансмиссия, как было доказано, благотворно влияет на познавательную способность, особенно на обучение и память. Было показано, что продолжительное лечение тезофенсином увеличивает уровень BDNF в головном мозге и может иметь антидепрессант эффект.
Клинические испытания
Фаза Результаты исследования IIB (TIPO-1), опубликованные в The Lancet, показали, что уровни потери веса за 6-месячный период были значительно выше, чем у любых доступных в настоящее время препаратов. Пациенты потеряли в среднем 12,8 кг при дозе 1 мг, 11,3 кг при дозе 0,5 мг и 6,7 кг при дозе 0,25 мг, по сравнению с потерей 2,2 кг в группе плацебо.
Всем участникам было рекомендовано соблюдать диету с дефицитом 300 ккал и постепенно увеличивать физическую активность до 30–60 минут в день. Средние потери веса за вычетом плацебо составили 4,5%, 9,2% и 10,6% в группах с дозами 0,25, 0,5 и 1 мг соответственно. Это примерно вдвое превышает потерю веса, которую вызывают лекарства, одобренные в настоящее время Управлением по контролю за продуктами и лекарствами США (FDA) для лечения ожирения.
NeuroSearch также сообщил о промежуточных результатах 48-недельного открытого расширенного исследования (TIPO-4), в котором 140 пациентов завершили 24-недельное исследование фазы IIB (TIPO -1) были повторно включены в исследование в среднем после 3-месячной отмывки. Первоначально все получали 0,5 мг тезофенсина один раз в сутки, но в первые 24 недели расширенного исследования было разрешено повышение дозы до 1,0 мг один раз в сутки. В этот момент все субъекты продолжали принимать дозу 0,5 мг в течение дополнительных 24 недель. 24-недельные промежуточные результаты для тех, кто ранее лечился тезофенсином 0,5 мг в TIPO-1, показали общую среднюю потерю веса от 13 кг до 14 кг за 48 недель лечения. Кроме того, TIPO-4 подтвердил результаты TIPO-1, поскольку пациенты, которые ранее лечились плацебо, потеряли примерно 9 кг за первые 24 недели исследования TIPO-4.
Побочные эффекты
В целом профиль безопасности тезофензина аналогичен профилю безопасности лекарств, одобренных в настоящее время для лечения ожирения. Наиболее частыми побочными эффектами у людей с ожирением были сухость во рту, головная боль, тошнота, бессонница, диарея и запор. Наблюдалась дозозависимая картина сухости во рту и бессонницы. Общий уровень отмены из-за нежелательных явлений в клинических испытаниях среди пациентов с ожирением составил 13% в группе тезофенсина и 6% в группе плацебо. Повышение артериального давления и частоты сердечных сокращений при применении терапевтически релевантных доз тезофенсина (0,25 мг и 0,5 мг) составляло 1-3 мм рт.ст. и до 8 ударов в минуту, соответственно.
По завершении фазы II клинических испытаний Saniona объявила что тезофенсин хорошо переносится с низкой частотой нежелательных явлений, низким увеличением частоты сердечных сокращений и отсутствием значительного влияния на артериальное давление.
Ссылки
From Wikipedia, the free encyclopedia
|
|
| Clinical data | |
|---|---|
| Pregnancy category |
|
| Routes of administration |
Oral |
| ATC code |
|
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Bioavailability | 90% |
| Metabolism | 15–20% renal; hepatic: CYP3A4 |
| Elimination half-life | 220 hours |
| Excretion | Not applicable |
| Identifiers | |
|
IUPAC name
|
|
| CAS Number |
|
| PubChem CID |
|
| ChemSpider |
|
| UNII |
|
| CompTox Dashboard (EPA) |
|
| Chemical and physical data | |
| Formula | C17H23Cl2NO |
| Molar mass | 328.28 g·mol−1 |
| 3D model (JSmol) |
|
|
SMILES
|
|
|
InChI
|
|
| |
Tesofensine (NS2330) is a serotonin–noradrenaline–dopamine reuptake inhibitor from the phenyltropane family of drugs, which is being developed for the treatment of obesity.[1] Tesofensine was originally developed by a Danish biotechnology company, NeuroSearch, who transferred the rights to Saniona in 2014.[2]
As of 2019, tesofensine has been discontinued for the treatment of Alzheimer’s and Parkinson’s disease but is in phase III clinical trial for obesity.[3]
History[edit]
Tesofensine was originally investigated for the treatment of Alzheimer’s disease and Parkinson’s disease,[4] and was subsequently dropped from development for these applications after early trial results showed limited efficacy for treatment of these diseases.[5][6] However, weight loss was consistently reported as an adverse event in the original studies, especially in overweight or obese patients.[7] Therefore, it was decided to pursue development of tesofensine for the treatment of obesity.
Tesofensine primarily acts as an appetite suppressant, but possibly also acts by increasing resting energy expenditure.[8] Phase II clinical trials for the treatment of obesity have been successfully completed.
Pharmacology[edit]
Metabolism and half-life[edit]
Tesofensine has a long half-life of about 9 days (220 h)[4] «and is mainly metabolized by cytochrome P4503A4 (CYP3A4) to its desalkyl metabolite M1» NS2360.[9][10] NS2360 is the only metabolite detectable in human plasma. It has a longer half-life than tesofensine, i.e. approximately 16 days (374 h) in humans, and has an exposure of 31–34% of the parent compound at steady state. In vivo data indicate that NS2360 is responsible for approximately 6% of the activity of tesofensine. As in animals, the kidney appears to play only a minor role in the clearance of tesofensine in humans (about 15–20%).
Transporter selectivity[edit]
Originally it had been reported that Tesofensine has IC50 of 8.0, 3.2 and 11.0nM at the DAT, NAT and 5HTT.[11] More recently, though, the following data was submitted: IC50 (nM) NE 1.7, SER 11, DA 6.5.[[12] cited in [13]] The revised IC50‘s would adequately explain the lack of efficacy in treating Parkinson’s disease, i.e. insufficient DRI potency relative to the SERT and the NET. This could also help account for why Tesofensine is not reliably self-administered by human stimulant abusers[14] since it has been believed to be the case that DAT inhibition is necessary for this and not NET inhibition.[15][16]
Tesofensine also indirectly potentiates cholinergic neurotransmission[17] proven to have beneficial effects on cognition, particularly in learning and memory. Sustained treatment with tesofensine has been shown to increase BDNF levels in the brain, and may possibly have an antidepressant effect.[12]
Clinical trials[edit]
Phase IIB trial (TIPO-1) results reported in The Lancet[18] showed levels of weight loss over a 6-month period that were significantly greater than those achieved with any currently available drugs. Patients lost an average of 12.8 kg on the 1 mg dose, 11.3 kg on the 0.5 mg dose and 6.7 kg on the 0.25 mg dose, compared with a 2.2 kg loss in the placebo group.
All participants were instructed to follow a diet with a 300 kcal deficit and to increase their physical activity gradually to 30–60 minutes of exercise per day. The placebo-subtracted mean weight losses were 4.5%, 9.2% and 10.6% in the 0.25 mg, 0.5 mg and 1 mg dose groups, respectively. This is approximately twice the weight loss produced by medications currently approved by the US Food and Drug Administration (FDA) for the treatment of obesity.
NeuroSearch has also reported interim results[8] from a 48-week, open-label, extension trial (TIPO-4) in which 140 patients who completed the 24-week phase IIB trial (TIPO-1) were re-enrolled after an average of 3 months’ wash-out. All were initially treated with 0.5 mg tesofensine once daily but up-titration to 1.0 mg once daily was allowed in the first 24 weeks of the extension study. At this time point, all subjects were continued on the 0.5 mg dose for an additional 24 weeks. The 24-week interim results for those who were previously treated with tesofensine 0.5 mg in TIPO-1 showed a total mean weight loss of between 13 kg and 14 kg over 48 weeks of treatment. Furthermore, TIPO-4 confirmed the TIPO-1 results since those patients who were previously treated with placebo lost approximately 9 kg in the first 24 weeks of the TIPO-4 study.
Adverse events[edit]
In general, the safety profile of tesofensine is similar to currently approved medications for the treatment of obesity. The most commonly reported side effects in the obese population were dry mouth, headache, nausea, insomnia, diarrhea and constipation. A dose-dependent pattern was observed for dry mouth and insomnia. The overall withdrawal rate due to adverse events in clinical trials in the obese population was 13% with tesofensine and 6% with placebo. Blood pressure and heart rate increases with the therapeutically relevant doses of tesofensine (0.25 mg and 0.5 mg) were 1–3 mmHg and up to 8 bpm, respectively.[8][18]
At the conclusion of phase II clinical trials, Saniona announced that tesofensine was well tolerated with low incidence of adverse events, low increase in heart rate and no significant effect on blood pressure.[19]
See also[edit]
- Hyperforin
- Phencyclidine
- Indatraline
- Dexanabinol
References[edit]
- ^ Doggrell SA. «Tesofensine – a novel potent weight loss medicine. Evaluation of: Astrup A, Breum L, Jensen TJ, Kroustrup JP, Larsen TM. Effect of tesofensine on bodyweight loss, body composition, and quality of life in obese patients: a randomised, double-blind, placebo-controlled trial. Lancet 2009 372; 1906–13″ Doggrell SA (July 2009). «Tesofensine—a novel potent weight loss medicine. Evaluation of: Astrup A, Breum L, Jensen TJ, Kroustrup JP, Larsen TM. Effect of tesofensine on bodyweight loss, body composition, and quality of life in obese patients: a randomised, double-blind, placebo-controlled trial. Lancet 2008;372:1906-13» (PDF). Expert Opinion on Investigational Drugs. 18 (7): 1043–6. doi:10.1517/13543780902967632. PMID 19548858. S2CID 207475155.
- ^ «NeuroSearch A/S signs agreement to transfer Phase I-II projects NS2359 and NS2330 (Tesofensine)». NeuroSearch company announcement. Retrieved 30 October 2014.
- ^ «Tesofensine — Saniona». AdisInsight. Springer Publishing. 29 January 2019. Retrieved 31 October 2019.
- ^ a b Bara-Jimenez W, Dimitrova T, Sherzai A, Favit A, Mouradian MM, Chase TN (October 2004). «Effect of monoamine reuptake inhibitor NS 2330 in advanced Parkinson’s disease». Movement Disorders. 19 (10): 1183–6. doi:10.1002/mds.20124. PMID 15390018. S2CID 21287239.
- ^ Hauser RA, Salin L, Juhel N, Konyago VL (February 2007). «Randomized trial of the triple monoamine reuptake inhibitor NS 2330 (tesofensine) in early Parkinson’s disease». Movement Disorders. 22 (3): 359–65. doi:10.1002/mds.21258. PMID 17149725. S2CID 37878251.
- ^ Rascol O, Poewe W, Lees A, Aristin M, Salin L, Juhel N, et al. (May 2008). «Tesofensine (NS 2330), a monoamine reuptake inhibitor, in patients with advanced Parkinson disease and motor fluctuations: the ADVANS Study». Archives of Neurology. 65 (5): 577–83. doi:10.1001/archneur.65.5.577. PMID 18474731.
- ^ Astrup A, Meier DH, Mikkelsen BO, Villumsen JS, Larsen TM (June 2008). «Weight loss produced by tesofensine in patients with Parkinson’s or Alzheimer’s disease». Obesity. 16 (6): 1363–9. doi:10.1038/oby.2008.56. PMID 18356831.
- ^ a b c NeuroSearch. «Tesofensine». http://www.neurosearch.dk/Default.aspx?ID=118 Accessed 17 May 2010.
- ^ Lehr T, Staab A, Tillmann C, Trommeshauser D, Raschig A, Schaefer HG, Kloft C (July 2007). «Population pharmacokinetic modelling of NS2330 (tesofensine) and its major metabolite in patients with Alzheimer’s disease». British Journal of Clinical Pharmacology. 64 (1): 36–48. doi:10.1111/j.1365-2125.2007.02855.x. PMC 2000606. PMID 17324246.
- ^ Lehr T, Staab A, Tillmann C, Nielsen EØ, Trommeshauser D, Schaefer HG, Kloft C (January 2008). «Contribution of the active metabolite M1 to the pharmacological activity of tesofensine in vivo: a pharmacokinetic-pharmacodynamic modelling approach». British Journal of Pharmacology. 153 (1): 164–74. doi:10.1038/sj.bjp.0707539. PMC 2199391. PMID 17982477.
- ^ Jorgen Scheel-Kruger, Peter Moldt, Frank Watjen. Tropane-derivatives, their preparation and use. U.S. Patent 6,288,079
- ^ a b Larsen MH, Rosenbrock H, Sams-Dodd F, Mikkelsen JD (January 2007). «Expression of brain derived neurotrophic factor, activity-regulated cytoskeleton protein mRNA, and enhancement of adult hippocampal neurogenesis in rats after sub-chronic and chronic treatment with the triple monoamine re-uptake inhibitor tesofensine». European Journal of Pharmacology. 555 (2–3): 115–21. doi:10.1016/j.ejphar.2006.10.029. PMID 17112503.
- ^ Marks DM, Pae CU, Patkar AA (December 2008). «Triple reuptake inhibitors: the next generation of antidepressants». Current Neuropharmacology. 6 (4): 338–43. doi:10.2174/157015908787386078. PMC 2701280. PMID 19587855.
- ^ Schoedel KA, Meier D, Chakraborty B, Manniche PM, Sellers EM (July 2010). «Subjective and objective effects of the novel triple reuptake inhibitor tesofensine in recreational stimulant users». Clinical Pharmacology and Therapeutics. 88 (1): 69–78. doi:10.1038/clpt.2010.67. PMID 20520602. S2CID 39849071.
- ^ Wee S, Wang Z, He R, Zhou J, Kozikowski AP, Woolverton WL (April 2006). «Role of the increased noradrenergic neurotransmission in drug self-administration». Drug and Alcohol Dependence. 82 (2): 151–7. doi:10.1016/j.drugalcdep.2005.09.002. PMID 16213110.
- ^ Wee S, Woolverton WL (September 2004). «Evaluation of the reinforcing effects of atomoxetine in monkeys: comparison to methylphenidate and desipramine». Drug and Alcohol Dependence. 75 (3): 271–6. doi:10.1016/j.drugalcdep.2004.03.010. PMID 15283948.
- ^ http://eprints.qut.edu.au/29667/1/c29667.pdf[bare URL PDF]
- ^ a b Astrup A, Madsbad S, Breum L, Jensen TJ, Kroustrup JP, Larsen TM (November 2008). «Effect of tesofensine on bodyweight loss, body composition, and quality of life in obese patients: a randomised, double-blind, placebo-controlled trial». Lancet. 372 (9653): 1906–1913. doi:10.1016/S0140-6736(08)61525-1. PMID 18950853. S2CID 30725634.
- ^ «Saniona’s tesofensine meets primary and secondary endpoints in Phase 3 obesity registration trial» (Press release). Saniona AB. GlobeNewswire. 17 December 2018. Retrieved 31 October 2019.
Тезофенсин
Tesofensine
Фармакологическое действие
Тезофенсин — ингибитор обратного захвата серотонина-норэпинефрина-дофамина (SNDRI).
Показания
Исследуется на предмет оценки фармакокинетики, эффективности, переносимости и безопасности при терапии ожирения.
Информация о действующем веществе Тезофенсин предназначена для медицинских и фармацевтических специалистов, исключительно в справочных целях. Инструкция не предназначена для замены профессиональной медицинской консультации, диагностики или лечения. Содержащаяся здесь информация может меняться с течением времени. Наиболее точные сведения о применении препаратов, содержащих активное вещество Тезофенсин, содержатся в инструкции производителя, прилагаемой к упаковке.
Медикаментозное лечение ожирения: прошлое, настоящее и будущее
Статья в формате PDF.
 А.С. Ларин
А.С. Ларин
Катастрофический рост распространенности ожирения в обществе свидетельствует о том, что с начала 80-х годов оно начало приобретать характер глобальной эпидемии. По оценкам специалистов, в настоящее время во всем мире около 1,6 млрд жителей имеют избыточную массу тела (индекс массы тела – ИМТ >25 кг/м2). За последние 40 лет (1975-2014) количество людей, страдающих ожирением, выросло со 105 до 641 млн человек, причем доля тучных мужчин за это время утроилась, а женщин – удвоилась. Согласно выводам исследователей, число людей в мире, страдающих от ожирения, превысило число жителей планеты, которые имеют недостаток массы тела. Стоимость расходов на ожирение и связанные с ним проблемы очень велики. В настоящее время только прямые расходы на ожирение в США превышают 100 млрд долларов. К сожалению, прогнозы на будущее пока неутешительны. Если сейчас в целом по всему миру около 36% людей имеют избыточную массу тела, а 23% населения нашей планеты страдают от ожирения, то ожидается, что в ближайшие два десятилетия число больных с избыточной массой тела увеличится еще в 2 раза. Согласно эпидемиологическим прогнозам, к 2025 г. избыточной массой тела и ожирением будут страдать 40% мужчин и 50% женщин. В связи с такими неутешительными данными ожирение признано ВОЗ новой неинфекционной пандемией XXI века [19, 22].
 С.М. Ткач
С.М. Ткач
У миллионов людей ожирение повышает риск серьезных медицинских проблем, таких как сахарный диабет (СД) 2 типа, метаболический синдром, кардиоваскулярные заболевания, неалкогольная жировая болезнь печени (НАЖБП), мышечно-скелетные расстройства, расстройства сна (синдром апноэ во сне), некоторые формы рака. Ожирение четко ассоциируется с повышением риска общей смертности (вследствие всех причин) [25].
 А.В. Пидаев
А.В. Пидаев
На сегодняшний день лечение ожирения включает несколько методов, объем которых зависит от выраженности избыточной массы тела: диетотерапию, режим физической активности, поведенческую терапию, назначение фармакологических препаратов (анорексигенного действия, блокаторы гидролиза и всасывания жиров и др.) и хирургические методы лечения. Вне зависимости от того, какой способ лечения выбран, все они направлены на то, чтобы помочь пациенту потреблять меньше калорий и увеличить его физическую активность с целью достижения оптимального состояния сердечно-сосудистой системы и контроля массы тела на протяжении длительного времени. Фармакотерапия рекомендована людям с ИМТ≥30 (или при ИМТ=27 с осложнениями), которые не в состоянии уменьшить массу тела только с помощью изменения образа жизни [5, 25].
Ниже в историческом плане рассмотрены основные медикаменты, которые в разное время применялись для лечения ожирения. Условно выделено 5 основных этапов применения различных препаратов: период до 1892 г., когда для лечения ожирения впервые были использованы тиреоидные гормоны; период с 1892 по 1940 г., когда были завершены клинические исследования амфетамина; период с 1940 по 1973 г., когда FDA впервые был утвержден фенфлурамин; период с 1973 по 1996 г., когда был открыт лептин и фенфлурамин был отозван с фармацевтического рынка; и, наконец, период с 1996 г. по настоящее время.
Препараты, применявшиеся до 1892 г.
Ожирение известно с древних времен, а монографии, в которых оно описано более подробно, берут начало, начиная с 18-го столетия. Использование медикаментов для лечения ожирения также имеет долгую историю, которая включает в себя использование «слабительных» и рвотных средств для увеличения потери «продуктов питания» через желудочно-кишечный тракт (ЖКТ). В один из наиболее широко применяемых медицинских трактатов 17-18 столетий входили такие средства для лечения ожирения, как табак, уксус, алоэ, корица, микстура из ревеня, слабительные микстуры, включавшие в разных сочетаниях тартар, корицу, имбирь, чеснок, лук-порей, семена руты и сахар [5].
Препараты, применявшиеся в период с 1892 по 1940 г.
В этот период применялись три основные группы препаратов – тиреоидные гормоны, динитрофенол и амфетамин.
Тиреоидные гормоны. У пациентов с гипотиреозом наблюдался набор массы тела по эндокринному типу, который хорошо поддавался лечению с помощью тиреоидного экстракта. Этот клинический эффект дал повод использовать тиреоидный экстракт для лечения ожирения у пациентов без гипотиреоза (впервые применено в 1892 г.). Хотя этот гормональный препарат имел последующие взлеты и падения в плане назначений, но до настоящего времени продолжает служить моделью препарата, который повышает метаболизм и расход энергии. Клиническое применение трийодтиронина, тироксина и тиреоидного экстракта было популярно, потому что провоцировало быструю потерю массы тела. К сожалению, этот эффект наблюдался в основном за счет снижения объема мышечной ткани, а не жировой [5].
Динитрофенол некоторое время использовался в лечении ожирения после того, как была отмечена потеря массы тела у работников заводов, на которых производился этот продукт, хотя это официально не было исследовано и ранее нигде не упоминалось [7].
Амфетамин был синтезирован в 1887 г. Поскольку препарат стимулировал бодрствование, эта его способность использовалась в лечении нарколепсии – состоянии повышенной сонливости. В 1937 г. в работах Nathanson было отмечено, что у 10 из 40 пациентов с нарколепсией отмечалось значительное снижение аппетита и потеря массы тела (на 3,2-9 кг) [24]. Одними из первых клинических исследований амфетамина как препарата, снижающего массу тела, были работы Lesses и Myerson. В исследовании принимали участие 17 пациентов с ожирением, которые находились на гипокалорийной диете (1400 кКал/сут) и принимали препарат в течение 2 недель. На протяжении времени, пока формировалась амфетаминовая зависимость, пациенты теряли в среднем 0,66 кг/неделю [23]. Было установлено, что амфетамин увеличивает концентрацию норэпинефрина и допамина в головном мозгу, причем норэпинефрин снижал аппетит, а допамин ассоциировался с развитием наркотической зависимости.
Препараты, применявшиеся между 1940 и 1973 гг.
К сожалению, после 2-й Мировой войны амфетамин и его производное метамфетамин из-за развития наркотической зависимости стали уличными наркотиками, что привело к поискам более безопасных альтернативных средств, которые подавляли голод, но не имели таких побочных эффектов. Для этого химиками-органиками были синтезированы три различные группы новых химических соединений.
Первая группа – подобные амфетамину симпатомиметики, которые уменьшали потребление пищи, но имели меньший или значительно меньший потенциал в плане развития зависимости, возможно, из-за преимущественного выделения норэпинефрина, а не допамина в головном мозге.
Вторая группа веществ – трициклические ингибиторы обратного захвата норэпинефрина, которые стали важной группой препаратов для лечения депрессии, но одновременно вызывали и снижение аппетита.
Третью группу препаратов представил фенфлурамин, способствующий высвобождению серотонина и частично блокирующий его обратный захват в нервных окончаниях. По своему действию фенфлурамин был схож с амфетамином, но не вызывал лекарственной зависимости. Синтез фенфлурамина дал абсолютно новый виток в развитии серотонинергических веществ как препаратов для лечения ожирения, хотя это и было ассоциировано с редкими случаями первичной легочной гипертензии [5, 25].
Препараты, применявшиеся между 1973 и 1996 гг.
В этот период были синтезированы дексфенфлурамин и эфедрин. Дексфенфлурамин – правовращающий изомер фенфлурамина, который также способен снижать аппетит и способствовать похудению. Двойное слепое рандомизированное мультицентрическое клиническое исследование Guy-Grand и соавт. в 1989 г., которое длилось в течение года, показало эффективность дексфенфлурамина в лечении ожирения, вследствие чего его применение было утверждено в 1996 г. FDA США [8]. К сожалению, в дальнейшем выяснилось, что этот препарат был кардиотоксичным и повышал риск развития первичной легочной гипертензии, вследствие чего он был отозван с рынка в сентябре 1997 г. и запрещен для клинического применения [14].
Эфедрин – симпатомиметик, который использовался для лечения бронхиальной астмы, но также повышал термогенез и снижал аппетит в клинических исследованиях. Самостоятельно этот препарат имел слабовыраженное действие, а в сочетании с кофеином представлял собой эффективную комбинацию для снижения массы тела [3].
Комбинированная терапия. В клиническом исследовании комбинированной терапии для лечения ожирения Weintraub и соавт. показали, что комбинация серотонинергического препарата (фенфлурамин) с адренергическим препаратом (фентермин) показала лучшую динамику в снижении массы тела с меньшими побочными эффектами, чем при лечении каждым препаратом отдельно. Кроме того, при наблюдении за больными более 3 лет во многих случаях отмечалось более длительное сохранение эффекта снижения массы тела. Информация о существенной потере массы тела в результате комбинации двух одобренных FDA препаратов – фентермина и фенфлурамина, получивших название Fen/Phen, быстро распространилась по всей стране и стала чрезвычайно популярной, хотя данная комбинация еще не была утверждена FDA США. Впервые тучные американцы стали выигрывать борьбу с лишней массой, что ранее случалось крайне редко. К сожалению, последующие длительные наблюдения выявили непредвиденные неблагоприятные последствия комбинации фенфлурамина с фентермином. В июле 1997 г. у пациентов, принимающих Fen/Phen, были задокументированы первые случаи клапанной болезни сердца [8]. После срочного повторного обсуждения эффектов указанной комбинации FDA заявила, что более чем у 30% пациентов, принимающих Fen/Phen, развивались клапанные пороки сердца. В связи с этим, 15 сентября 1997 г. комбинация фенфлурамина и фентермина (но не фентермин) была изъята из продаж во всем мире и запрещена для клинического применения [5, 14].
Комбинации эфедрина с кофеином или растительные комбинации алкалоидов эфедры с растительным кофеином стали второй эффективной и популярной комбинацией препаратов для снижения массы тела [3, 5]. В клинических исследованиях Astrup и соавт. была показана эффективность сочетания эфедрина и кофеина, а общественный энтузиазм в лечении растительными комбинациями, которые были доступны без рецепта, привел к их широкому распространению. В последующем появились сообщения, свидетельствующие о повышении риска смерти при их применении, что привело к большому общественному скандалу и прекращению использования эфедрина для снижения массы тела. Эти два примера отзыва ранее зарегистрированных препаратов явились не первыми и не последними случаями, которые привели к запрещению препаратов для лечения ожирения после их длительного изучения (табл. 1).

Препараты, исследовавшиеся и/или применяющиеся после 1996 г.
Начиная с 1996 г., начался самый плодотворный период в исследовании препаратов для лечения ожирения. Сначала для клинического применения был утвержден дексфенфлурамин, а двумя годами позже в исследованиях на мышах с генетическим ожирением был идентифицирован лептин.
Лептин. Было установлено, что у животных и людей с ожирением, у которых отмечался недостаток этой молекулы, при лечении лептином наблюдалась реверсия ожирения. Механизм действия синтезирующегося в жировой ткани лептина заключается в передаче сигналов в ЦНС, контролирующих потребление пищи через рецепторы меланокортина, которые модулируются нейропептидом-Y, меланоцитстимулирующим гормоном и меланин-концентрирующим гормоном [18].
К сожалению, препараты, которые были синтезированы на основе этих знаний, до сих пор не оправдали своих ожиданий. Так, в проведенных клинических исследованиях введение лептина людям с ожирением оказывало только незначительные эффекты на массу тела. Такой побочный эффект как локальное раздражение в местах инъекций ограничил использование препарата. В настоящее время лептин утвержден FDA США для лечения липодистрофий.
После 1996 г. при ожирении также изучалась эффективность нескольких препаратов – аналогов нейропептидов, но они, к сожалению, до конца не оправдали тех надежд, которые на них возлагали.
Антагонисты рецепторов нейропептида-Y. Нейропептид-Y – широко распространенный нейропептид, который действует на 5 рецепторов – Y‑1, Y‑2, Y‑4, Y‑5 и Y‑6. Он стимулирует аппетит, снижает расход энергии и повышает массу тела путем активации Y‑1 и Y‑5 рецепторов в гипоталамусе. Было проведено несколько клинических исследований селективного антагониста рецептора Y‑5. В одном из исследований наблюдалась существенная потеря массы тела, что доказало влияние антагониста рецептора Y на регуляцию массы. К сожалению, второе исследование показало незначительный эффект, который был не достаточен для продолжения последующих исследований [10].
Аксокин – модифицированная форма реснитчатого нейтрофильного фактора, которая действует посредством янус-киназной сигнальной системы, используя билептин. Он снижал аппетит у животных с недостатком лептина или лептиновых рецепторов, а в дозозависимом исследовании показал существенный клинический ответ со снижением массы тела на 3-5%. К сожалению, у 70% принимавших этот препарат, развивался иммунный ответ с высоким титром антител к препарату, что резко снижало его эффективность [5].
Холецистокинин (ХЦК) – пептид, который может оказывать влияние на ЖКТ и ЦНС. ХЦК снижает аппетит как у людей, так и у животных. Были разработаны и протестированы аналоги пептида, но никаких клинических данных опубликовано не было [16].
Пептид YY (PYY3-36) – другой кишечный пептид, который вырабатывается L-клетками. При его интраназальном применении потребление калорий во время приема пищи снижалось на 30% у 12 субъектов с ожирением и на 29% – у худых пациентов. В клинике пока не применяется [5].
Оксинтомодулин – кишечный пептид, схожий с PYY и ХЦК, вырабатывается в ЖКТ и может снижать аппетит. В исследовании здоровых волонтеров с ожирением и повышенной массой тела оксинтомодулин, который вводился подкожно, снижал массу тела на 2,3±0,4 кг в сравнении с 0,5±0,5 кг в контрольной группе в финале исследования. В клинике пока не применяется [25].
Антагонисты грелина. Грелин – гастроинтестинальный пептид, который стимулирует аппетит. В настоящее время проводятся исследования по изучению его эффективности при ожирении, однако клинических данных пока нет.
Таким образом, представленные выше нейропептиды продемонстрировали весьма скромные результаты, а физиологически важные пути контроля потребления пищи оказались далекими от реальной клинической практики и утверждения на их основе новых методов терапии ожирения.
Тем не менее в этот период было разработано и протестировано много других препаратов, в первую очередь, центрального действия, воздействующих на обмен серотонина и допамина, хотя только некоторые из них были утверждены для лечения ожирения.
Сибутрамин (Меридия) – центральный высокоселективный ингибитор обратного захвата норадреналина и серотонина, и, в меньшей степени, допамина, приводящий к подавлению аппетита. Он снижает аппетит на 23% в течение 1-й недели, и на 26% – после двух недель применения, стимулируя тем самым потерю массы тела [15]. Сибутрамин также эффективен в предупреждении рецидива набора массы тела, как было показано в клиническом исследовании STORM (Sibutramine Trial of Obesity Reduction and Maintenance), которое длилось 2 года [20]. Сибутрамин был одобрен FDA еще в 1997 г. и широко применялся во всем мире для лечения ожирения. Применение сибутрамина ограничивали его частые побочные эффекты, такие как сухость во рту, головная боль, запоры и инсомния. Кроме того, сибутрамин приводил к тахикардии и повышению АД, а также нес в себе риск серотонинового синдрома, если применялся вместе с ингибиторами моноаминоксидазы (МАО), триптанами и опиоидами. В качестве противопоказаний к применению сибутрамина рассматривались плохо контролируемая гипертензия, ишемическая болезнь сердца, аритмии, сердечная недостаточность, инсульт, эпилепсия, тяжелые заболевания почек и печени, применение ингибиторов МАО. Учитывая такой неблагоприятный спектр побочных эффектов и противопоказаний, а также данные последних исследований, свидетельствующих о том, что его применение повышает кардиоваскулярный риск, в частности, риск возникновения инфарктов миокарда и инсультов, FDA потребовала прекратить маркетинг сибутрамина [21]. Поэтому в октябре 2010 г компания-производитель сибутрамина объявила о добровольном изъятии этого препарата с рынка США и в настоящее время по клиническим показаниям для лечения ожирения он больше не применяется. Ранее сибутрамин уже был отозван с рынков Европы и Канады.
Еще один препарат с таким же механизмом действия (тезофензин) принимал участие в 6-месячном исследовании, также вызывал побочные действия на сердечно-сосудистую систему и поэтому не был рекомендован для клинического применения [15].
Флюоксетин – селективный ингибитор обратного захвата серотонина, был исследован для лечения ожирения, но рецидив набора массы тела на протяжении второго полугодия привел к прекращению дальнейшего тестирования препарата по этим показаниям [11]. В настоящее время широко применяется как антидепрессант.
Бупропион, другой антидепрессант и ингибитор обратного захвата норэпинефрина и допамина, в клинических исследованиях в дозах 300 и 400 мг/сут снижал массу тела на 6,2 и 7,2% соответственно [2]. В настоящее время входит в состав комбинированного препарата Contrave® (налтрексон/бупропион) для лечения ожирения.
Экопипам – антагонист первого и пятого допаминовых рецепторов, изучался для лечения кокаиновой зависимости. Он также находился в разработке в качестве препарата для лечения ожирения, но исследование было прекращено [5].
Топирамат – противоэпилептический препарат, при назначении которого отмечалась потеря массы тела на 3,9-7,3%. В 6-месячном плацебо-контролируемом дозозависимом исследовании наблюдались 385 человек, разделенных на 5 групп по дозировкам препарата – 64 мг/сут, 94 мг/сут, 192 мг/сут, 384 мг/сут и плацебо, при этом отмечалась дозозависимая потеря массы тела, хотя были и побочные эффекты в виде парестезий, сонливости, ухудшения концентрации внимания и памяти [5, 25]. В настоящее время применяется для лечения ожирения в виде комбинации фентермин/топирамат (Qsymia®).
Римонабант блокирует каннабиноидные рецепторы 1 типа, находящиеся почти во всех тканях организма, включая участки мозга, отвечающие за насыщение, адипоциты и ЖКТ. Каннабиноидные рецепторы начали исследовать после того, как было замечено, что марихуана и гашиш, содержащие тетрагидроканнабинол (основное психотропное вещество), повышают аппетит, особенно в отношении сладкого. Исследования показали, что римонабант, разработанный как блокатор этих рецепторов, уменьшал аппетит. На периферии он тормозил отложение жира и увеличивал выделение адипонектина, чем уменьшал проявления инсулинорезистентности. Эффективность римонабанта была достаточно высока – потеря массы тела на 8,6-8,8 кг по сравнению с плацебо (2,2-2,6 кг) по результатам трех масштабных исследований, проведенных в Европе [9]. К сожалению, в связи с тем, что он воздействует на центр удовольствия, то при приеме римонабанта наблюдались нарушения настроения, в частности, депрессия и склонность к суицидам, нервозность, бессонница. В связи с этим в 2008-2009 гг. римонабант был запрещен к применению и изъят из продаж.
Современные препараты, утвержденные для лечения ожирения
Согласно существующим рекомендациям FDA США, прием всех новых лекарств для лечения ожирения в течение года должен обеспечивать потерю массы тела на 5% больше по сравнению с плацебо или обеспечивать >5% потери массы тела не менее чем у 35% пациентов. При этом очень желателен благоприятный метаболический профиль и низкая частота побочных эффектов. FDA очень тщательно отслеживает безопасность всех новых препаратов, поскольку их в течение продолжительного времени будет принимать большое количество людей. Так как связанные собственно с ожирением риски реализуются в течение длительного времени, стандартом безопасности для всех новых лекарств становится соотношение риск/польза.
Все утвержденные FDA США препараты для лечения ожирения (табл. 2) можно разделить на 2 группы [5]. Первая группа – препараты, утвержденные для долгосрочной терапии ожирения, включают в себя орлистат, лоркасерин, комбинацим фентермин/топирамат (Qsymia®) и налтрексон/бупропион (Contrave®), а также лираглутид. Как показала большая часть долговременных клинических испытаний, эти лекарства значительно увеличивают потерю массы тела по сравнению с плацебо, причем максимальная потеря массы достигается в период между 20-28 неделями применения и в среднем составляет 8-10% (с плацебо – 4-6%). Снижение массы тела наблюдается до тех пор, пока используется препарат. Вторая группа – симпатомиметики, утвержденные FDA США для краткосрочной терапии (обычно <12 недель), включают диэтилпропион, фентермин, бензфетамин и фендиметразин.
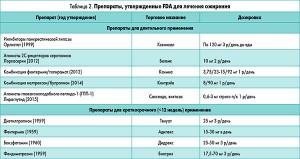
Препараты, утвержденные для длительного лечения ожирения
Орлистат (Ксеникал) – препарат периферического действия, не обладающий системными эффектами. Фармакологическое действие препарата обусловлено его способностью инактивировать липазу ЖКТ, что препятствует расщеплению и последующему всасыванию около 30% жиров. В результате возникает хронический дефицит энергии, что способствует снижению массы тела. Чтобы орлистат оказывал свое действие, диета должна состоять из жиров не менее чем на 30%. Также следует учитывать, что при низкожировой диете орлистат практически не работает. Он назначается по 120 мг 3 р/день во время или в течение часа после еды при условии наличия жиров в пище [6].
Данный препарат был зарегистрирован в США в 1999 г. и доказал свою эффективность по сравнению с плацебо. По данным различных исследований, потеря массы тела при приеме орлистата составила от 8,8 кг (плацебо – 5,8 кг) до 3,3 кг (плацебо – 1,3 кг). Что касается побочных эффектов, то системных зарегистрировано почти не было, поскольку препарат работает только в просвете кишечника. Основными побочными эффектами являются симптомы отсутствия всасывания жира, который проходит транзитом и попадает из тонкой кишки в толстую, где его в норме не должно быть совсем, что вызывает нарушения пищеварения в виде так называемого жирного кала, вплоть до его недержания, а также сильное газообразование в кишечнике.
Это налагает определенные ограничения на пациентов, особенно в плане отдыха, выхода на улицу и т.д. Также в некоторых случаях были замечены нарушения всасывания жирорастворимых витаминов (А, Е и К), поэтому, чтобы не было необходимости следить за витаминным балансом всех пациентов, рекомендуется назначать поливитамины на фоне приема орлистата.
В комбинации с умеренно гипокалорийной диетой препарат значительно уменьшает массу тела, препятствует повторной прибавке массы, улучшает течение сопутствующих заболеваний и повышает качество жизни. Орлистат рекомендован для длительного контроля массы тела у больных ожирением. Противопоказаниями к назначению являются: синдром мальабсорбции, гиперчувствительность к препарату или его компонентам.
Лоркасерин (Белвик) – агонист серотониновых 2С-рецепторов. Серотониновые рецепторы являются хорошо известной мишенью для уменьшения приема пищи (по крайней мере, в исследованиях на животных) и усиления расхода энергии. На них было направлено действие фенфлурамина, который был отозван с рынка в 1997 г. из-за его способности вызывать вальвулопатию (с последней ассоциируется серотониновый рецептор 2В). Принцип действия лоркасерина заключается в активации серотониновых рецепторов головного мозга типа 2С, что способствует более быстрому насыщению даже при небольшом количестве принятой пищи. Препарат блокирует чувство голода и это позволяет пациенту потерять в среднем 5% массы тела. Результат довольно-таки скромный, однако эксперты FDA отметили, что применение Белвика не вызывает серьезных побочных эффектов в отличие от других препаратов. Потеря массы тела при приеме лоркасерина в двух больших плацебо-контролируемых исследованиях была умеренной и составила 4,8 и 4%, что приближается к требованиям FDA [26].
Лоркасерин хорошо переносится. Наиболее распространенные побочные действия – головная боль, тошнота, головокружение, слабость, сухость во рту, запоры – выражены слабо и проходят довольно быстро. Препарат нельзя принимать вместе с ингибиторами обратного захвата серотонина и ингибиторами МАО из-за риска развития серотонинового синдрома.
Фентермин/топирамат продленного действия (Ксимиа) – комбинация двух известных препаратов. Первый относится к группе симпатомиметиков – стимуляторов ЦНС (его механизм сходен с сибутрамином) и применяется для снижения массы тела, второй – для лечения эпилепсии и мигрени. Данная комбинация изучалась в 3 дозовых режимах, в каждом из которых дозы препаратов были ниже стандартных, применяемых при монотерапии. Степень потери массы тела при приеме данной комбинации в течение года была наиболее высокой и оказалась на 12% выше чем плацебо. Поскольку топирамат является ингибитором карбоангидразы, возникали побочные эффекты – нарушение вкуса и чувствительности, а также покалывание в руках и вокруг рта. Кроме того, отмечены сухость во рту, запоры, инсомния и нарушения зрения при применении высоких доз.
Несмотря на отсутствие серьезных побочных эффектов, отчет при комбинации фентермин/топирамат FDA на своем заседании в октябре 2010 г. не утвердил. Однако завершившееся в 2011 г. рандомизированное двойное слепое контролируемое исследование показало хорошие результаты, в связи с чем в 2012 г. эта комбинация была утверждена для длительного лечения ожирения. Фентермин снижает аппетит путем повышения норэпинефрина в гипоталамусе, а топирамат снижает аппетит, влияя на GABA-рецепторы [1].
Бупропион/налтрексон (Контрэйв) – комбинация препаратов, каждый из которых известен своей эффективностью в снижении массы тела. Антагонист опиоидных рецептров налтрексон применяется для лечения опиатной и алкогольной зависимостей, а схожий по структуре с психостимулятором катиноном антидепрессант бупропион применяется также для терапии депрессии и облегчения отказа от курения. Концепция данной комбинации заключается в том, что количественно регулируемая супрессия эндорфинов под влиянием бупропиона может быть ингибирована налтрексоном. Для исследования эффективности и безопасности препарата Контрэйв было привлечено порядка 4,5 тыс. пациентов, имеющих лишнюю массу тела. Часть из них получала лекарственное средство, остальные принимали плацебо. Также участники должны были вести здоровый образ жизни, подразумевающий соблюдение низкокалорийной диеты и физическую активность. Оказалось, что из числа пациентов, принимавших экспериментальный препарат и не страдающих СД, 42% удалось уменьшить массу тела минимум на 5%, при этом в контрольной группе их было 17%. Из числа участников, которые страдали СД 2 типа, 36% пациентов смогли потерять минимум 5% своей массы, в группе плацебо – 18%. Профиль побочных эффектов включал тошноту, рвоту, головную боль, кожный зуд, запор и диарею [13].
Препарат Контрэйв одобрен для использования взрослыми пациентами с ИМТ >30 в качестве средства для лечения ожирения, а также для пациентов с ИМТ >27 в качестве средства для терапии избыточной массы тела. Основным условием для назначения препарата является наличие у пациентов хотя бы одного заболевания, связанного с лишней массой тела – гипертензии, СД 2 типа или повышенного уровня холестерина.
Лираглутид (Саксенда, Виктоза) – представляет собой аналог человеческого глюкагоноподобного пептида‑1 (ГПП‑1), произведенный методом биотехнологии рекомбинантной ДНК с использованием штамма Saccharomyces cerevisiae, имеющий 97% гомологичности аминокислотной последовательности эндогенному человеческому ГПП‑1. Лираглутид связывается и активирует рецептор ГПП‑1 (ГПП‑1Р), устойчив к метаболическому распаду, период его полувыведения из плазмы после подкожного введения составляет 13 ч. Лираглутид уменьшает массу тела у человека преимущественно посредством уменьшения массы жировой ткани. Он не увеличивает 24-часовой расход энергии, а уменьшение массы тела происходит за счет уменьшения потребления пищи. Препарат регулирует аппетит с помощью усиления чувства наполнения желудка и насыщения, одновременно ослабляя чувство голода и уменьшая предполагаемое потребление пищи. Лираглутид также стимулирует секрецию инсулина и уменьшает неоправданно высокую секрецию глюкагона глюкозозависимым образом, а также улучшает функцию β-клеток поджелудочной железы, что приводит к снижению концентрации глюкозы натощак и после приема пищи. Мультицентровое европейское исследование ежедневных инъекций лираглутида (1,2, 1,8, 2,4 или 3 мг) показало снижение массы тела на 4,8, 5,5, 6,3 и 7,2 кг соответственно в сравнении с потерей на 2,8 кг в группе плацебо и 4,1 кг у пациентов при лечении орлистатом [4]. Ранее лираглутид был утвержден FDA и European Medicines Agency в качестве сахароснижающего препарата в дозе 1,8 мг/сут, а в 2015 г. он получил одобрение FDA в качестве средства для лечения ожирения у взрослых пациентов с минимум одним сопутствующим заболеванием, например, СД 2 типа либо сердечно-сосудистыми нарушениями. Препарат должен приниматься в сочетании с низкокалорийной диетой и физическими упражнениями. Лираглутид не рекомендуется использовать пациентам с личной либо семейной историей медуллярной карциномы щитовидной железы или с синдромом эндокринной неоплазии второго типа.
Препараты, утвержденные для кратковременного лечения ожирения
Симпатомиметики, такие как диэтилпропион, фентермин, бензфетамин и фендиметразин объединены в одну группу, потому что действуют как норэпинефрин и были протестированы до 1973 г. Согласно классификации U.S. Drug Enforcement Agency, диэтилпропион и фентермин отнесены к препаратам IV класса, а бензфетамин и фендиметразин – к препаратам III класса, которые считаются потенциально вызывающими зависимость, хотя эта вероятность достаточно мала. Тем не менее все эти препараты утверждены для краткосрочного приема (<12 недель) [5, 25].
Фентермин. Хотя этот препарат был утвержден FDA еще в 1959 г., он остается наиболее часто назначаемым для снижения массы тела в США. Так как фентермин используется только в качестве краткосрочного препарата, информации о его долгосрочном применении в виде монотерапии нет. По данным корейского исследования, 12-недельный прием фентермина в дозе 30 мг/сут привел к потере массы тела на 8,1±3,9 кг (в группе плацебо – 1,7±2,9 кг) [1, 8]. Следует помнить, что все симпатомиметики провоцируют центральное возбуждение, манифестирующее в качестве инсомнии, нервозности и сухости во рту. Этот эффект более явный в начале приема препарата и постепенно уменьшается при продолжении приема.
Симпатомиметики также могут провоцировать тахикардию и артериальную гипертензию, в связи с чем, они с осторожностью назначаются пациентам из группы риска сердечно-сосудистых заболеваний и не рекомендуются пациентам с ИБС в анамнезе.
Препараты для лечения других заболеваний, способствующие потере массы тела
Как известно, побочным эффектом многих препаратов является либо набор, либо потеря массы тела. Поэтому рациональным выбором для лечения пациента с ожирением и сопутствующим заболеванием, например, СД 2 типа или депрессией, является назначение сахароснижающего препарата или антидепрессанта, способствующего потере массы тела [12]. В таблице 3 перечислены препараты для лечения СД, артериальной гипертензии, гиперхолестеринемии и психотических расстройств, которые способны приводить к потере или набору массы тела.
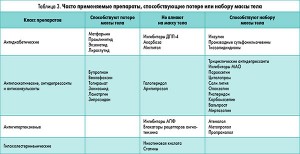
Говоря о лечении ожирения с сопутствующим СД, следует иметь в виду, что инсулин способствует набору массы тела от 1,8 до 6,6 кг. Два наиболее широко применяемых препарата сульфонилмочевины (глипизид и глибенкламид) также стимулировали набор массы тела в большинстве исследований в диапазоне 0,3-4 кг. Препараты тиазолидиндионов, такие как розиглитазон и пиоглитазон спосбствовали набору массы тела в диапазоне 0,18-1,5 кг и более. Остальные препараты нейтральны по отношению к массе тела или способствуют ее снижению [5, 12].
Большой интерес с точки зрения возможного применения для терапии ожирения, особенно при наличии нарушений углеводного обмена, представляет метформин, повышающий печеночную и периферическую чувствительность к эндогенному инсулину, не действуя на его секрецию. Препарат замедляет всасывание углеводов в ЖКТ, а также снижает аппетит. В связи с этим терапия метформином сопровождается уменьшением или стабилизацией массы тела, а также снижением отложения висцерального жира. Метформин также снижает или предотвращает набор массы тела во время лечения антипсихолитиками. Важно заметить, что метформин обладает кардиоваскулярным протективным эффектом, связанным с гиполипидемическим и антиатерогенным действием препарата, его благоприятным влиянием на липидный обмен (снижает на 10-30% окисление свободных жирных кислот), эндотелиальную функцию, сосудистую реактивность, систему гемостаза и реологию крови, в частности за счет уменьшения гиперкоагуляции и гиперактивности тромбоцитов. Лечение препаратом начинают с дозы 500-850 мг, принимаемой во время ужина или на ночь. В дальнейшем суточная доза препарата увеличивается на 500-850 мг каждые 1-2 недели. Максимальная рекомендованная доза для больных с ожирением составляет 1500-1700 мг/сут в режиме 2-3 приема [5].
Прамлинитид – модифицированная форма амилина – пептида, секретируемого совместно с инсулином β-клетками поджелудочной железы. Прамлинитид утвержден FDA для лечения диабета, но он также снижает и массу тела [5, 25].
Экзенатид – пептид, продуцируемый слюнными железами ящериц, на 53% сходный с GLP‑1, но имеющий более продолжительный период полураспада. Препарат был утвержден в качестве лечения СД 2 типа, который не контролировался приемом метформина и препаратов сульфонилмочевины. Экзенатид снижает уровни глюкозы натощак и через 2 ч после еды, замедляет опустошение желудка и снижает аппетит, благодаря чему умеренно снижается масса тела. Побочные эффекты – головная боль, тошнота и рвота, которые уменьшаются после снижения дозы. Потеря массы тела на экзенатиде наблюдалась без модификации образа жизни – диеты и физических нагрузок [5, 25].
При наличии у пациентов с избыточной массой тела депрессии препаратами выбора могут быть бупропион и флюоксетин. Бупропион может также использоваться с целью сокращения или предупреждения увеличения массы тела у людей, пытающихся бросить курить [2, 11].
Будущее в развитии фармакотерапии ожирения
К сожалению, достигнув больших успехов в понимании механизмов развития ожирения и кратковременного контроля за аппетитом, у нас до сих пор нет принципиально новых эффективных препаратов. Провал клинических применений лептина, агониста нейропептида-Y и агониста меланокортинового рецептора произошел вследствие нежелательных побочных эффектов или недостаточной эффективности. Тем не менее все-таки некоторые механизмы действия препаратов для лечения ожирения в перспективе кажутся многообещающими [5].
Первый механизм – действие препаратов на ЖКТ. В дополнение к достаточно хорошо изученной энтеро-эндокринной системе мы знаем, что в ЖКТ имеются вкусовые и нюхательные рецепторы, которые потенциально могут стать терапевтической мишенью в контроле за аппетитом.
Второй механизм – воздействие сразу на несколько мишеней. Здесь можно провести аналогию между ожирением и гипертензией. Известно, что лечение гипертензии имеет целый арсенал надежных препаратов, которые работают посредством различных механизмов. Тем не менее монотерапия гипертензии очень часто неэффективна, поэтому в настоящее время почти повсеместно используются комбинации препаратов. Эта стратегия начала использоваться и в лечении ожирения, в частности она уже нашла свое выражение в утверждении комбинаций фентермин/топирамат и бупропион/налтрексон [17].
Еще один механизм связан с повышением АД при приеме некоторых эффективных в лечении ожирения препаратов. Как уже указывалось выше, сибутрамин, ингибитор обратного захвата серотонина и норэпинефрина, был изъят из обращения вследствие повышения кардиоваскулярных рисков и АД. Другой препарат – тезофензин, который показал снижение массы тела более чем на 10% в клинических исследованиях, также повышал АД. Поэтому понимание тесной взаимосвязи АД и снижения массы тела может в дальнейшем помочь в разработках новых видов лечения.
Наконец, согласно экспериментальным данным, лептин все-таки способствует полной реверсии ожирения, поэтому изучение эффектов и терапевтического применения лептина или его аналогов будет разрабатываться и в дальнейшем. Так, уже начал изучаться новый препарат – белоранид, снижающий у пациентов с редкой формой ожирения – синдромом Прадер-Вилли, количество жировой ткани на 8,1% в сравнении с контролем [5, 25].
Выводы
Текущий подход к фармакотерпии пациентов с ожирением очень часто подразумевает отсроченное назначение препаратов до развития сопутствующей патологии и лечение уже развившихся осложнений, хотя потеря массы тела сама по себе может смягчить многие из этих сопутствующих заболеваний или осложнений. На сегодняшний день основной целью фармакотерапии ожирения является изменение поведенческих привычек, которые в дальнейшем приведут к изменению стиля жизни. Для этого применяются утвержденные FDA и рассмотренные выше препараты, предназначенные для долгосрочного применения и снижения массы тела. В настоящее время сохраняются надежды на возможность эффективного лечения ожирения до развития осложнений. В качестве будущих стратегий рассматривается более широкое применение комбинаций препаратов для коррекции массы тела и сахароснижающих препаратов с эффектами снижения массы тела.
Безусловно, следует учитывать уроки прошлого. Во-первых, к ожирению следует относиться как к хроническому заболеванию, поэтому препараты должны назначаться с долгосрочной перспективой, до появления стабильного эффекта. Во-вторых, важно до конца понять механизмы, посредством которых препараты снижают массу тела, при этом сами медикаменты должны назначаться в составе комплексной терапии, включая модификацию диеты и физической активности. В-третьих, поскольку мы уже имеем печальный опыт в развитии непредвиденных неблагоприятных отдаленных последствий некоторых препаратов, ключевыми факторами уже известных и новых разрабатываемых лекарственных средств должны быть их безопасность и переносимость. Поскольку отдаленные последствия фармакотерапии в будущем предугадать и предупредить достаточно сложно, преимущество в фармакотерапии ожирения должны иметь пациенты с патологическим ожирением, которые будут получать от этого наибольшую пользу. Назначение лекарственной терапии для снижения массы тела с косметической целью требует более высокого уровня безопасности по сравнению с тем, который есть на сегодняшний день.
Литература
1. Allison D., Gadde K., Garwey W. Controlled-release phentermine/topiramat in severe obese adult. Obesity Silver Spring 2012; 20 (2): 330-42.
2. Anderson J., Greenway F., Fujioka K. Bupropion SR enchanced weight loss: 40-week double-blind, placebo-controlled trial. Obese Res 2002; 10 (7): 633-41.
3. Astrup A., Breum L., Toubro S. The effect and safety of an ephedrine/caffeine compound compare to ephedrine, caffeine and placebo in obese subjects on an energy restricted diet. Int J Obes Relat Metab Disord 1992; 16 (4): 269-77.
4. Astrup A., Rossner S., Van Gaal. Effects of liraglutide in the treatment of obesity. Lancet 2009; 374: 1606-16.
5. Bray G.A. Medical treatment of obesity: the past, present and the future. Clinical gastroenterology 2014; 28 (4): 665-684.
6. Chanoine J., Hampl S., Bodrin M. Effect of orlistat on weight and body composition in obese adolescents. J Av Med Assoc 2005; 293 (23): 2873-83.
7. Сolman E. Dinitrophenol and obesity: an early twentieth-century regulatory dilemma. Regul Toxicol Pharmacol 2007; 48 (2): 115-7.
8. Connally H., Crary J., McGoon M. Valvular heart disease associated with fenfluramine/phenthermine. New Engl J Med 1997; 337 (9): 581-8.
9. Desppres J., Golay A., Sostrom L. Effect of rimonabant on metabolic risk factors in overweight patients with dyslipidemia. N Engl J Med 2005; 353 (20): 2121-34.
10. Erondu N., Gantz I., Musser B. Neyropeptide Y5 receptor antagonist does not induce clinically meaningful weight loss in overweith and obese adults. Cell Metab 2006; 4 (4): 275-82.
11. Goldstein D., Rampey J., Roback H. Efficacy and safety of long-term fluoxetine treatment of obesity-maximizing effect. Obese Res 1995; 3 (4): 481-90.
12. Greenway F., Whitehouse M, Guttadauria M. Rational design of combination medication for the treatment of obesity. Obesity Silver Spring 2009; 17 (1): 30-9.
13. Greenway F., Fudjioka K., Plodkovski R. Effect of naltrexone plus bupropion on weight loss in overweight and obese adults. Lancet 2010; 376: 595-605.
14. Guy-Grand B., Apfelbaum M., Crepaldi G. Unternational trial of long-term dexfenfluramine in obesity. Lancet 1989; 2 (8672): 1142-5.
15. Halford J., Harroid J., Boiland I. Serotonergic drugs: effect on appetite expression and use for the treatment of obesity. Drugs 2007; 67 (1): 27-55.
16. Heil G., Ross S. Chemical agents affecting appetite/ In Bray G., editor. Obesity in perspective. Fogarty International Center Series on Preventive Med; 1976: p. 429-40.
17. Hendricks E., Rothman R., Greenway F. How physician obesity specialists use drugs to treat obesity. Obesity Silver Spring 2009; 17: 1730-5.
18. Heymsfield S., Greenberg A., Fudjioca R. Recombinant leptin for weight loss in obese and lean adults. J Am Med Assoc 1999; 282 (16): 1568-75.
19. Hill J.O., Wyatt H.R., Reed G.W., Peters J.C. Obesity and the environment: where do we go from here? Science 2003; 299 (5608): 853-5.
20. James W., Astrup A., Finer N. Effect of sibutramine on weight maintenance after weith loss: a randomized study. STORM study group. Lancet 2000; 356 (9248): 219-25.
21. James W., Caterson I., Coutinho W. Effect of sibutramine on cardiovascular outcomes in overweight and obese subjects. N Engl J Med 2010; 263 (10): 905-17.
22. Kumanyika S.K., Obarzanek E. Pathways to obesity prevention. Report of a National Institutes of Health Workshop. Obes Res 2003;11 (10):1263-74.
23. Lesses M., Myerson A. Human autonomic pharmacology. N Engl J Med 1938; 218 (3): 119-24.
24. Nathanson M. The central action of betaaminopropylbenzene. J Am Mad Assoc 1937; 108 (7): 528-31.
25. Rucker D., Padwal R., Curioni S., Lau D. Long term pharmacotherapy for obesity and overweight: updated meta-analysis. BMJ 2007; 335 (7631): 1194-9.
26. Smith S., Wessman N., Anderson C. Multicenter placebo-controlled trial of lorcaserin for weight management. N Engl J Med 2010; 363 (3): 2450-56.
Статья впервые опубликована в журнале
«Клінічна ендокринологія та ендокринна хірургія»,
№ 1, 2017 г.
- Номер:
- Тематичний номер «Діабетологія, Тиреоїдологія, Метаболічні розлади» № 2 (38), травень 2017 р.
СТАТТІ ЗА ТЕМОЮ Ендокринологія
10.07.2023
Ендокринологія
Вплив добавок вітаміну D та селену на перебіг автоімунного захворювання щитоподібної залози – хвороби Хашимото
Хвороба Хашимото (ХХ) – це автоімунне захворювання щитоподібної залози (АЗЩЗ), пов’язане з її гіпофункцією, лімфоцитарною інфільтрацією та підвищенням титрів антитиреоїдних антитіл, особливо до тиреоглобуліну (АТ-TГ) і тиреопероксидази (АТ-ТПО). ХХ – це Т-лімфоцит-опосередкований імунний розлад із постійним ушкодженням ЩЗ, зумовленим взаємодією факторів довкілля з певними генами у сприйнятливих пацієнтів. Одними з таких факторів є рівні вітаміну D (VD), йоду та селену (Se) [1]. Представлений у статті огляд літератури базується на електронному пошуку в базі даних PubMed і Google Scholar, публікацій із січня 2011 по березень 2019 року щодо пацієнтів із ХХ, які отримували вищезазначені добавки окремо або в комбінації з іншими в рамках клінічних досліджень. В огляді описано важливість і функції цих мікронутрієнтів і наведено результати досліджень.
…
10.07.2023
Терапія та сімейна медицина
Ендокринологія
Оновлення рекомендацій ADA 2023: основні акценти
У грудні 2022 року Американська діабетична асоціація (ADA) опублікувала оновлені «Стандарти надання медичної допомоги при цукровому діабеті 2023 р.». У них було враховано результати найсучасніших наукових і клінічних досліджень. До Стандартів увійшли важливі практичні рекомендації з менеджменту діабету і предіабету, у тому числі щодо: діагностики цукрового діабету (ЦД) 1 типу, ЦД 2 типу та гестаційного діабету в молоді та дорослих; стратегії профілактики або відстрочки ЦД 2 типу; терапевтичних підходів, які можуть зменшити кількість ускладнень, знизити серцево-судинні (СС) та ниркові ризики та поліпшити якість життя пацієнтів. Ще одне ключове оновлення стосується місця агоністів рецепторів глюкагоноподібного пептиду‑1 (арГПП‑1) в терапії ЦД, які наразі рекомендовано як першу ін’єкційну терапію для пацієнтів із ЦД 2 типу.
…